| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://jnr.elmerpub.com |
Case Report
Volume 16, Number 1, March 2026, pages 49-54

Lhermitte-Duclos Disease and Neurofibromatosis Type 1 in a Patient With a Follow-Up of More Than Twenty Years
Rohan Hublikara, Garrett Gianneschib, c ![]() , Janet Elgallabb
, Janet Elgallabb
aNew Jersey Medical School, Rutgers University, New Jersey, NJ, USA
bNeurology Department, Child Neurology Division, Rutgers-New Jersey Medical School, Newark, NJ, USA
cCorresponding Author: Garrett Gianneschi, Neurology Department, Child Neurology Division, Rutgers-New Jersey Medical School, Newark, NJ, USA
Manuscript submitted September 25, 2025, accepted February 14, 2026, published online March 13, 2026
Short title: LDD and NF-1: More Than 20 Years of Follow-Up
doi: https://doi.org/10.14740/jnr1053
| Abstract | ▴Top |
Lhermitte-Duclos disease (LDD) is a rare condition, with approximately 300 cases reported. We present over 20 years of the natural history of a young woman with the rare coexistence of LDD and neurofibromatosis type 1 (NF-1), as well as intractable epilepsy, infantile spasms, and cognitive and motor impairments. The cerebellar gangliocytoma remained stable on neuroimaging from early childhood into adulthood, while subclinical electroencephalogram (EEG) abnormalities improved during early adolescence but persisted into adulthood. This case highlights the importance of interdisciplinary care and close monitoring for evolving neuropsychiatric, developmental, and ophthalmological manifestations. The investigation and outcome identify novel combinations of electroencephalographic patterns, genetic abnormalities, and clinical phenotype in LDD and NF-1, offering valuable insights into the complex interplay of these rare conditions.
Keywords: Lhermitte-Duclos disease; Dysplastic cerebellar gangliocytoma; NF-1; D/EE-SWAS; Infantile spasm; Intractable epilepsy; HCN2 channelopathy
| Introduction | ▴Top |
Lhermitte-Duclos disease (LDD), also known as dysplastic cerebellar gangliocytoma, is a benign, hamartomatous lesion of the cerebellum, with a pathognomonic “tiger-striped” appearance on T1-weighted magnetic resonance imaging (MRI). There are approximately 300 reported cases, and it is most often associated with Cowden syndrome (CS) and mutations in the PTEN gene [1]. Neurofibromatosis type 1 (NF-1) is a neurocutaneous disorder characterized by the formation of neurofibromas and peripheral nerve tumors, with a wide range of clinical manifestations including cutaneous, skeletal, ophthalmological, and neurological features. Patients with both disorders appear to be exceedingly rare, with only one other such patient found to be reported in the literature [2]. In this case, the patient’s electronic medical record from the time of her childhood diagnosis to adulthood was available for review. As such, the aim of this > 20-year longitudinal case report is to highlight the evolution of clinical manifestations, as well as electroencephalographic, radiological and laboratory findings in a patient with these rare comorbidities to inform diagnosis, prognostication, and management of similar cases in the future.
| Case Report | ▴Top |
We present the case of a woman in her 20s with LDD in combination with NF-1, intractable epilepsy, and intellectual impairment. She was diagnosed with NF-1 at birth due to cutaneous findings of cafe au lait macules, a facial plexiform neurofibroma of the left upper lip, and axillary freckling. Her mother was also noted to have multiple cutaneous findings consistent with NF-1. The patient later presented with infantile spasms (IS) and was successfully treated with adrenocorticotropic hormone and topiramate. In early childhood she again developed seizures multiple times per day, consisting of eye blinking, screaming, and loss of consciousness. Subsequent magnetic resonance imaging (MRI) revealed a dysplastic cerebellar gangliocytoma involving the left cerebellum and midbrain, consistent with LDD, which co-occurred with electroencephalogram (EEG) abnormalities that were topographically concordant with the lesion.
Despite reportedly meeting developmental milestones in infancy, by early childhood the patient was noted to have gross motor, fine motor, and speech delays which persisted into adolescence. Her history and neurological exam have shown subtle but persistent findings of clumsiness, dysmetria and mild difficulties with tandem gait since early childhood, with few changes into adulthood. Her seizure semiology continued to evolve over her lifetime; by middle childhood, her seizures were 3-min episodes of staring, followed by full body atony and right arm stiffening. By early adolescence, they presented as generalized tonic–clonic seizures occurring a few times per year, mostly due to difficulty obtaining medication or complying with her anti-seizure medication (ASM) regimen.
The patient also developed complications of NF-1 during her clinical course. In infancy, she was diagnosed with right esotropia and lateral nystagmus. During early childhood, she underwent bilateral medial rectus recession surgery, which improved but did not fully correct her strabismus. Ophthalmological evaluation revealed Lisch nodules, bilateral myopic astigmatism, and pre-glaucoma. During early adolescence, she developed a new internuclear ophthalmoplegia (INO), which did not correspond to new findings on neuroimaging or ophthalmological exam and subsequently resolved without intervention. She was also noted in middle childhood to have enlarged cup-disc ratios with high risk for glaucoma; she is followed biannually for glaucoma screening but has not required treatment.
The patient was also diagnosed with attention deficit hyperactivity disorder (ADHD) in childhood due to extensive behavioral problems at school and home, including distractibility, hyperexcitability, and emotional dysregulation. She was trialed on various doses of methylphenidate and amphetamine salts with only minor effect, and these medications were discontinued during early adolescence. In her adolescence, her ADHD appeared to improve significantly, allowing her to focus on tasks and function semi-independently. However, she remained notably distractible on interview and at home, requiring frequent redirection during conversation and having difficulty with most activities of daily living (ADLs). She was able to complete secondary school with special education services, enrolled in extended schooling to age 21, and planned to continue with vocational rehabilitation services after schooling.
Investigations
The patient initially received neuroimaging in early childhood with MRI brain without intravenous contrast, which showed a 2.0 × 2.3 cm homogeneous, nonenhancing mass-like lesion expanding the left cerebellum, causing mass effect on the fourth ventricle (Fig. 1). Subsequent neuroimaging with MRI brain with and without intravenous contrast every 1–3 years demonstrated essentially unchanged size of the cerebellar mass with increasing age.
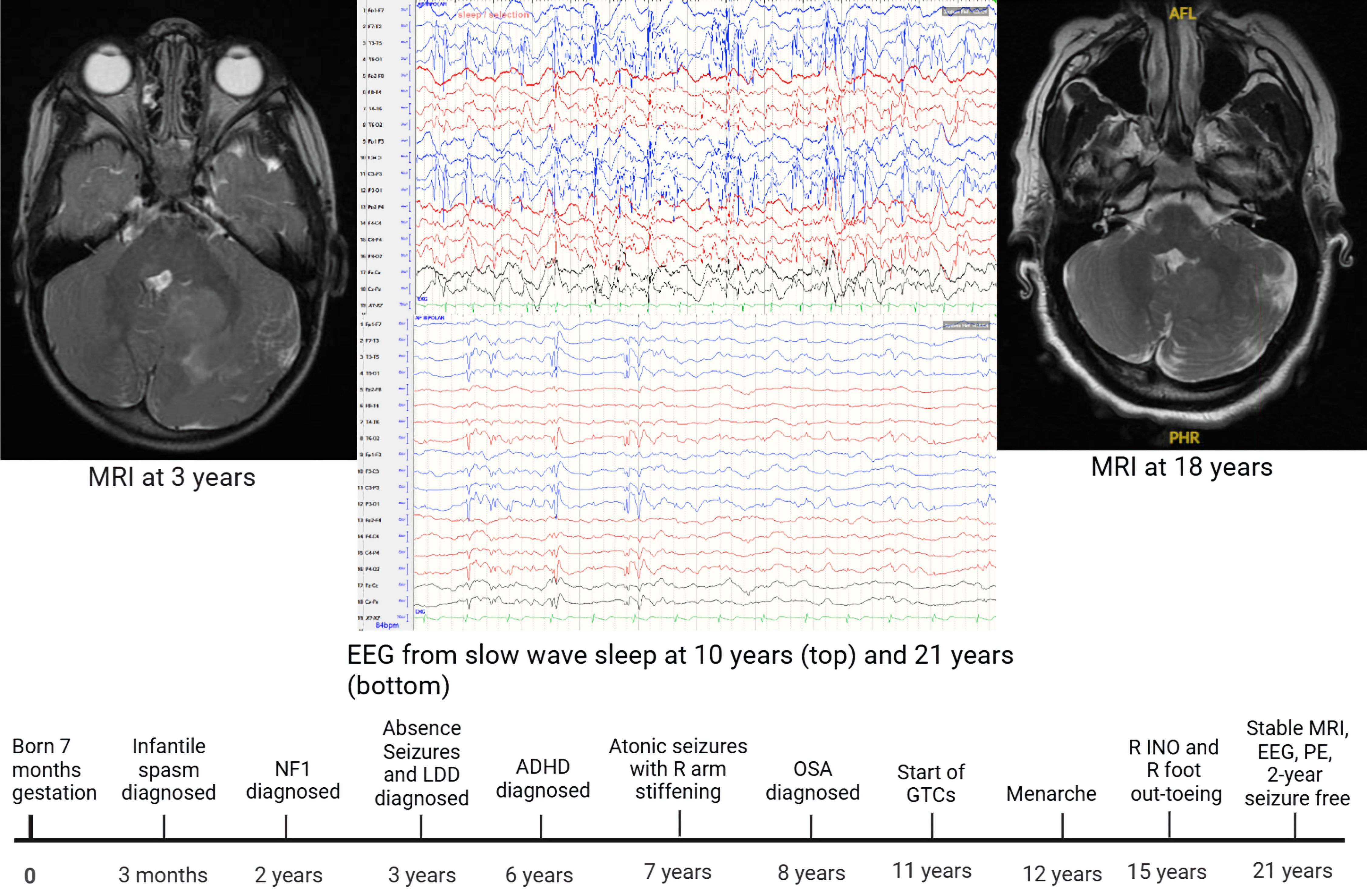 Click for large image | Figure 1. Timeline showing notable events throughout patient’s lifetime (bottom). Stable MRI findings at age 3 years (left) and 18 years (right) show stable gangliocytoma and patent fourth ventricle. EEG during slow-wave sleep at age 10 years (top middle) shows near-continuous spike–wave, and at age 21 years (bottom middle) shows significantly reduced spike–wave frequency in posterior left leads. LDD: Lhermitte-Duclos disease; NF-1: neurofibromatosis type 1; MRI: magnetic resonance imaging; EEG: electroencephalogram; INO: internuclear ophthalmoplegia; OSA: obstructive sleep apnea; GTCs: generalized tonic–clonic seizures; PE: physical examination. |
Due to her history of seizures, the patient has also undergone multiple EEGs throughout her life. Initial EEG findings in early childhood showed continuous rhythmic spike and wave activity from left occipital–posterior temporal region and focal slowing. Across serial EEGs from toddlerhood through late adolescence, epileptiform activity maximal over the left occipital–posterior temporal region progressively decreased. In toddlerhood, discharges were near-continuous during both wakefulness and non-rapid eye movement (NREM) sleep, with a sleep spike–wave index (SWI) > 85%. In the setting of documented developmental impairment, these findings supported a diagnosis of developmental and/or epileptic encephalopathy with spike–wave activation in sleep (D/EE-SWAS) per International League Against Epilepsy (ILAE) criteria. By late adolescence, the epileptiform burden had declined to frequent but intermittent activity during wakefulness (about 60% of wake epochs) and was less prominent in NREM sleep (< 45% of sleep epochs), consistent with attenuation of the prior D/EE-SWAS pattern with residual focal epileptiform activity. (Fig. 1).
While being followed by pediatric ophthalmology and neuro-ophthalmology, the patient received serial tonometry to assess intraocular pressure. The patient also received an optical coherence tomography (OCT) scan, which revealed possible right temporal thinning with no other abnormal findings. Subsequent visual examinations were stable, and further imaging was not required.
Genetic testing showed no PTEN mutation. A heterozygous mutation in the NF1 gene (variant pAsn2364Thrfs*) was identified and classified as likely pathogenic. Two heterozygous mutations in the SRCAP gene (variants p.Ala2404Val and p.Pro2461Ala) were detected and classified as variants of uncertain significance. A homozygous mutation in the HCN2 gene (variant p.Gln32Asnfs*230) was identified and classified as likely pathogenic. In addition, a heterozygous mutation in the KAT6B gene (variant p.Gly1698Ser) was detected and classified as a variant of uncertain significance.
Treatment
The patient was treated for seizures throughout her life. Her IS were managed with adrenocorticotropic hormone (ACTH) and topiramate. Her ASM regimen was expanded over her childhood from valproate alone to include levetiracetam, then lamotrigine, with dose titrations due to breakthrough seizures and increasing age. Her current ASM regimen is as follows: valproate, 500 mg by mouth, twice daily; levetiracetam, 750 mg by mouth, twice daily; and lamotrigine, 100 mg by mouth, twice daily.
Additionally, the patient received early intervention as a toddler for developmental delay. In early childhood, she received special education services which continued throughout her schooling, consisting of one-to-one instruction and reduced class sizes. She also received speech and occupational therapy for a lisp and fine motor delay respectively.
Finally, for ADHD, the patient was treated with dextroamphetamine, 10–20 mg (dose increased with age) by mouth once a day throughout early and middle childhood, which was discontinued by middle-childhood through shared decision-making with the patient’s parents.
Follow-up and outcomes
The patient’s overall outcome has been favorable. She has been seizure-free for more than 2 years, with prior breakthrough seizures related mostly to difficulty in obtaining medications. Her primary side effect from this medication regimen is weight gain and she has otherwise tolerated her medications well. Her mother was granted guardianship over her, and she was enrolled in extended schooling after completing secondary school. She has limited independence, has not attempted to enter the workforce, and is not able to maintain independent living due to intellectual impairment. However, she has continued to expand her ADLs and instrumental activities of daily living (IADLs) and is continuing with vocational rehabilitation services following completion of extended schooling. She continues to receive surveillance MRIs every 2–3 years for her dysplastic cerebellar gangliocytoma, as well as serial EEGs every 1–2 years to monitor her subclinical electrographic activity.
Patient’s perspective
The patient’s mother relates that she is thankful for the progress her child has made. Before age of 14, the patient was not able to express herself, was significantly impacted by behavioral issues, and her mother thought she “wasn’t going to make it.” Then, at 14 years old, “it was like a light went on,” and the patient became interactive, responsive, and seemed to “become her own person.”
| Discussion | ▴Top |
LDD is an infrequently described disease with a varied presentation. Age of onset varies from infancy to the fifth or sixth decade of life; some sources suggest comorbidity with CS is exclusive to adult-onset disease (> 18 years), while others question this association [1, 3–6]. Clinical presentations vary from subtle cerebellar deficits to headaches and herniation due to mass effect. Strabismus and nystagmus are also reported, usually due to compression of cranial nerves [1]. Treatment has consisted of sub-total or gross surgical resection versus a wait-and-see approach, and successful resection is often curative [1, 3]. To our knowledge, comorbidity of LDD with NF-1 has only been reported in a single other case, and IS have not been reported in association with LDD, except in a previous paper describing our patient [2, 7]. IS, while rare, may occur with increased frequency in NF-1, and associations between NF-1 and ADHD, epilepsy, strabismus, glaucoma, and intellectual disability, are well described in the literature [8, 9].
The electrographic pattern of continuous spike and wave activity during slow sleep, as described by Bhat et al [7] in our patient, has not otherwise been reported in association with LDD or NF-1 but is associated with significant neuropsychosocial impairment and varied seizure presentations that generally remit by age 15 [10, 11]. However, in this patient with a pathogenic NF1 variant and LDD (dysplastic cerebellar gangliocytoma), the longitudinal EEG evolution supports a sleep-activated epileptiform encephalopathy phenotype that is best framed within the ILAE construct of D/EE-SWAS. Serial recordings demonstrated a strikingly high early-life epileptiform burden—near-continuous spike/polyspike activity maximal over the left occipital–posterior temporal region with sleep SWI > 85%—followed by progressive attenuation across development, with late-adolescent studies showing reduced NREM involvement (< 45%) and residual frequent wake discharges (about 60% of wake epochs). This trajectory is consistent with the recognized natural history of SWAS-spectrum disorders, in which the sleep-activated component may diminish over time while focal epileptiformity persists. Importantly, the consistent occipital–posterior temporal predominance and associated focal slowing argue for a supratentorial epileptogenic network rather than direct epileptogenicity of the cerebellar lesion; thus, any relationship between dysplastic cerebellar gangliocytoma and the encephalopathy phenotype should be described as associative. Given the known association of NF1 with epilepsy and structural substrates for focal epilepsy, this case highlights the need to interpret SWAS patterns in the broader context of underlying neurocutaneous disease, evolving developmental course, and regional EEG physiology when attributing mechanism and counseling prognosis.
Seizures are a rare manifestation of LDD and have been described in patients with and without CS; in LDD, seizures have been successfully managed with surgical resection and medical management alone, although both treatment methods have also failed to achieve seizure freedom in some patients [2, 12, 13].
Though our patient is currently undergoing further evaluation of her genetic panel, mutations in NF-1 and HCN2 are preliminarily thought likely to have clinical significance and have the American College of Medical Genetics and Genomics (ACMG) classifications of “likely pathogenic.” Mutations involving the NF-1 gene are well known to cause NF-1 in an autosomal dominant fashion with varied clinical presentations, although the pathogenic variant in our patient has not been described previously [14]. The patient’s homozygous (biallelic) pathogenic variant mutation in HCN2 is novel in 1kG and is a frameshift deletion in exon 1, predicted to cause loss of function through protein truncation. HCN2 mutations have been documented in a limited number of individuals, with phenotypes including intellectual disability, generalized tonic–clonic epilepsy, absence seizures, and developmental/epileptic encephalopathy; however, many disease-causing variants appear to be gain-of-function mutations. Of the few reported loss-of-function pathogenic variants, known phenotypes include intellectual disability with and without epilepsy. Notably, in a study including 12 individuals with loss-of-function variants, 8/8 subjects with biallelic variants had severe intellectual disability, contrasting with our patient’s mild-moderate intellectual disability [15, 16]. While SRCAP mutations have often been associated with Floating-Harbor syndrome (FHS), causing skeletal abnormalities not seen in our patient, mutations outside the FHS locus may be associated with mild intellectual disability and ADHD, though the clinical significance of the SRCAP mutation in our patient is unknown [17]. KAT6B mutations, in contrast, have been associated with Say-Barber-Biesecker syndrome (SBBS) and genitopatellar syndrome (GPS), characterized respectively by facial abnormalities and hypoplastic patellae, neither of which describe our patient [18].
This more than 20-year longitudinal case report describes a unique constellation of clinical, genetic, electroencephalographic, and radiologic findings. The patient’s epilepsy and intellectual disability have evolved over childhood with overall improvement, but the relative contributions of her underlying conditions remain difficult to disentangle. Both NF-1 and LDD have been reported in association with epilepsy, and although neuroimaging has not demonstrated an additional gross supratentorial abnormality, other mechanisms (including subtle cortical malformation, remote network effects, or sequelae of early-life epileptic encephalopathy such as IS) cannot be excluded. While correlations among her epilepsy syndrome, neurodevelopmental profile, and cerebellar lesion cannot be proven, several convergent findings suggest a focal cortical epileptogenic network: her persistent EEG abnormalities localize to the left occipital–posterior temporal region, and serial EEGs in early life demonstrated a markedly sleep-activated epileptiform burden consistent with D/EE-SWAS, which attenuated with age while leaving residual focal epileptiform activity. These data support a supratentorial localization and warrant caution in attributing epileptogenicity directly to the cerebellar lesion; at most, any relationship between the dysplastic cerebellar gangliocytoma and epilepsy should be framed as associative, particularly in the absence of seizure remission temporally linked to lesion-directed therapy. Her broader genetic findings (even without a PTEN variant) highlight the potential value of comprehensive genetic evaluation in patients with LDD to refine diagnostic framing and guide anticipatory management.
Finally, we would like to highlight that our patient experienced marked improvement in her independence and intellectual function over her childhood and adolescence and has achieved seizure control via medical management alone. While the underlying cause(s) of our patient’s epilepsy and intellectual disability are unclear due to her multiple complex neurological conditions, the patient’s current outcome may assist in prognostication for patients with similar presentations.
Learning points
Dysplastic cerebellar gangliocytoma is a benign, slow-growing tumor which may present in childhood with or without other features of CS.
Dysplastic cerebellar gangliocytoma has been rarely reported in association with epilepsy and, in uncommon cases, with sleep-activated epileptiform patterns consistent with diagnosis of spike–wave activation in sleep, though a causal link has not been demonstrated.
Childhood LDD may be successfully managed with serial observation alone, even in a patient with multiple neurological comorbidities including intractable epilepsy.
In a patient with LDD, NF-1, intellectual disability and intractable epilepsy of uncertain cause, medical management alone was effective in achieving seizure control.
Whole gene sequencing in patients with multiple neurological comorbidities including LDD may reveal genetic abnormalities distinct from those found in CS.
Acknowledgments
We are grateful to the patient and family for permitting long-term clinical care and documentation.
Financial Disclosure
The authors received no financial support for the research, authorship, or publication of this article.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Informed Consent was obtained from the mother of the child for sharing longitudinal clinical data, imaging, EEG, and genetic information.
Author Contributions
Rohan Hublikar, MS4: primary author, neurological data synthesis and manuscript drafting. Garrett Gianneschi, DO: radiologic interpretation, clinical data compilation, and manuscript drafting. Janet Elgallab, MD: supervision, manuscript editing, and critical review.
Data Availability
The authors declare that all relevant clinical data supporting the findings of this study are included in the manuscript. Additional information may be made available upon reasonable request, in accordance with institutional policy and patient confidentiality.
Abbreviations
LDD: Lhermitte-Duclos disease; NF-1: neurofibromatosis type 1; CS: Cowden syndrome; MRI: magnetic resonance imaging; EEG: electroencephalogram; SWI: spike–wave index; D/EE-SWAS: developmental and/or epileptic encephalopathy with spike–wave activation in sleep; ILAE: International League Against Epilepsy; ASM: anti-seizure medication; INO: internuclear ophthalmoplegia; ADHD: attention deficit hyperactivity disorder; ADLs: activities of daily living; OCT: optical coherence tomography; IS: infantile spasms; FHS: Floating-Harbor syndrome; SBBS: Say-Barber-Biesecker syndrome; GPS: genitopatellar syndrome
| References | ▴Top |
- Alanazi AI, Alanezi T, Aljofan ZF, Alarabi A, Elwatidy S. Lhermitte-Duclos disease: a systematic review. Surg Neurol Int. 2023;14:351.
doi pubmed - Yesildag A, Baykal B, Ayata A, Kerman G, Koroglu M, Olgar S, Oyar O. Lhermitte-Duclos disease associated with neurofibromatosis type-1 and non-ossifying fibroma. Acta Radiol. 2005;46(1):97-100.
doi pubmed - Wang Q, Zhang S, Cheng J, Liu W, Hui X. Lhermitte-Duclos disease: clinical study with long-term follow-up in a single institution. Clin Neurol Neurosurg. 2017;162:53-58.
doi pubmed - Khandpur U, Huntoon K, Smith-Cohn M, Shaw A, Elder JB. Bilateral recurrent dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) in Cowden syndrome: a case report and literature review. World Neurosurg. 2019;127:319-325.
doi pubmed - Zak M, Ledbetter M, Maertens P. Infantile Lhermitte-Duclos disease treated successfully with rapamycin. J Child Neurol. 2017;32(3):322-326.
doi pubmed - Magana M, Landeta-Sa AP, Lopez-Flores Y. Cowden disease: a review. Am J Dermatopathol. 2022;44(10):705-717.
doi pubmed - Bhat S, Ming X, Dekermenjian R, Chokroverty S. Continuous spike and wave in slow-wave sleep in a patient with Rett syndrome and in a patient with Lhermitte-Duclos syndrome and neurofibromatosis 1. J Child Neurol. 2014;29(12):NP176-180.
doi pubmed - Bernardo P, Cinalli G, Santoro C. Epilepsy in NF1: a systematic review of the literature. Childs Nerv Syst. 2020;36(10):2333-2350.
doi pubmed - Hirabaru K, Matsuo M. Neurological comorbidity in children with neurofibromatosis type 1. Pediatr Int. 2018;60(1):70-75.
doi pubmed - Posar A, Visconti P. Continuous spike-waves during slow sleep today: an update. Children (Basel). 2024;11(2):169.
doi pubmed - Panayiotopoulos CP. In: The epilepsies: seizures, syndromes and management. Oxfordshire (UK), 2005.
pubmed - Robinson S, Cohen AR. Cowden disease and Lhermitte-Duclos disease: an update. case report and review of the literature. Neurosurg Focus. 2006;20(1):E6.
doi pubmed - Assi J, Chyta M, Mavridis I. Lhermitte-Duclos disease with concomitant KCNT2 gene mutation: report of an extremely rare combination. Childs Nerv Syst. 2023;39(11):3295-3299.
doi pubmed - Ece Solmaz A, Isik E, Atik T, Ozkinay F, Onay H. Mutation spectrum of the NF1 gene and genotype-phenotype correlations in Turkish patients: Seventeen novel pathogenic variants. Clin Neurol Neurosurg. 2021;208:106884.
doi pubmed - Kessi M, Peng J, Duan H, He H, Chen B, Xiong J, Wang Y, et al. The Contribution of HCN channelopathies in different epileptic syndromes, mechanisms, modulators, and potential treatment targets: a systematic review. Front Mol Neurosci. 2022;15:807202.
doi pubmed - Houdayer C, Phillips AM, Chabbert M, Bourreau J, Maroofian R, Houlden H, Richards K, et al. HCN2-associated neurodevelopmental disorders: data from patients and xenopus cell models. Ann Neurol. 2025;98(3):573-589.
doi pubmed - Rots D, Chater-Diehl E, Dingemans AJM, Goodman SJ, Siu MT, Cytrynbaum C, Choufani S, et al. Truncating SRCAP variants outside the Floating-Harbor syndrome locus cause a distinct neurodevelopmental disorder with a specific DNA methylation signature. Am J Hum Genet. 2021;108(6):1053-1068.
doi pubmed - Gannon T, Perveen R, Schlecht H, Ramsden S, Anderson B, Kerr B, Day R, et al. Further delineation of the KAT6B molecular and phenotypic spectrum. Eur J Hum Genet. 2015;23(9):1165-1170.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, including commercial use, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.