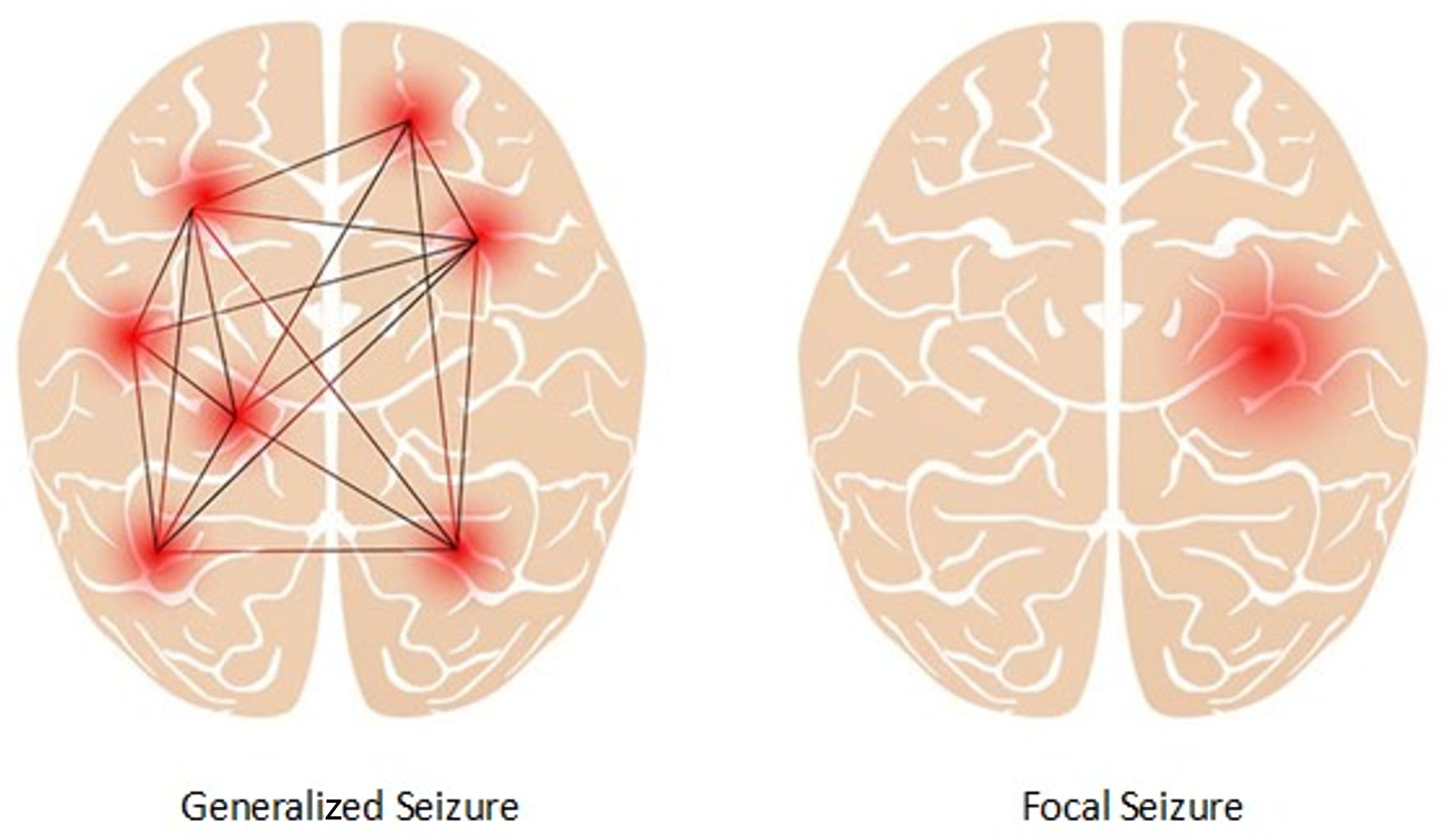
↓ Figure 1. Focal seizure and generalized
seizure.
| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://jnr.elmerpub.com |
Review
Volume 15, Number 4, December 2025, pages 149-164
Psychoeducational Therapy and Non-Pharmacological Therapeutic Intervention Approach for Pediatric Epilepsy Neuropsychiatric Comorbidities: A Neuroscience-Informed Strategy for Seizure Management
Figures
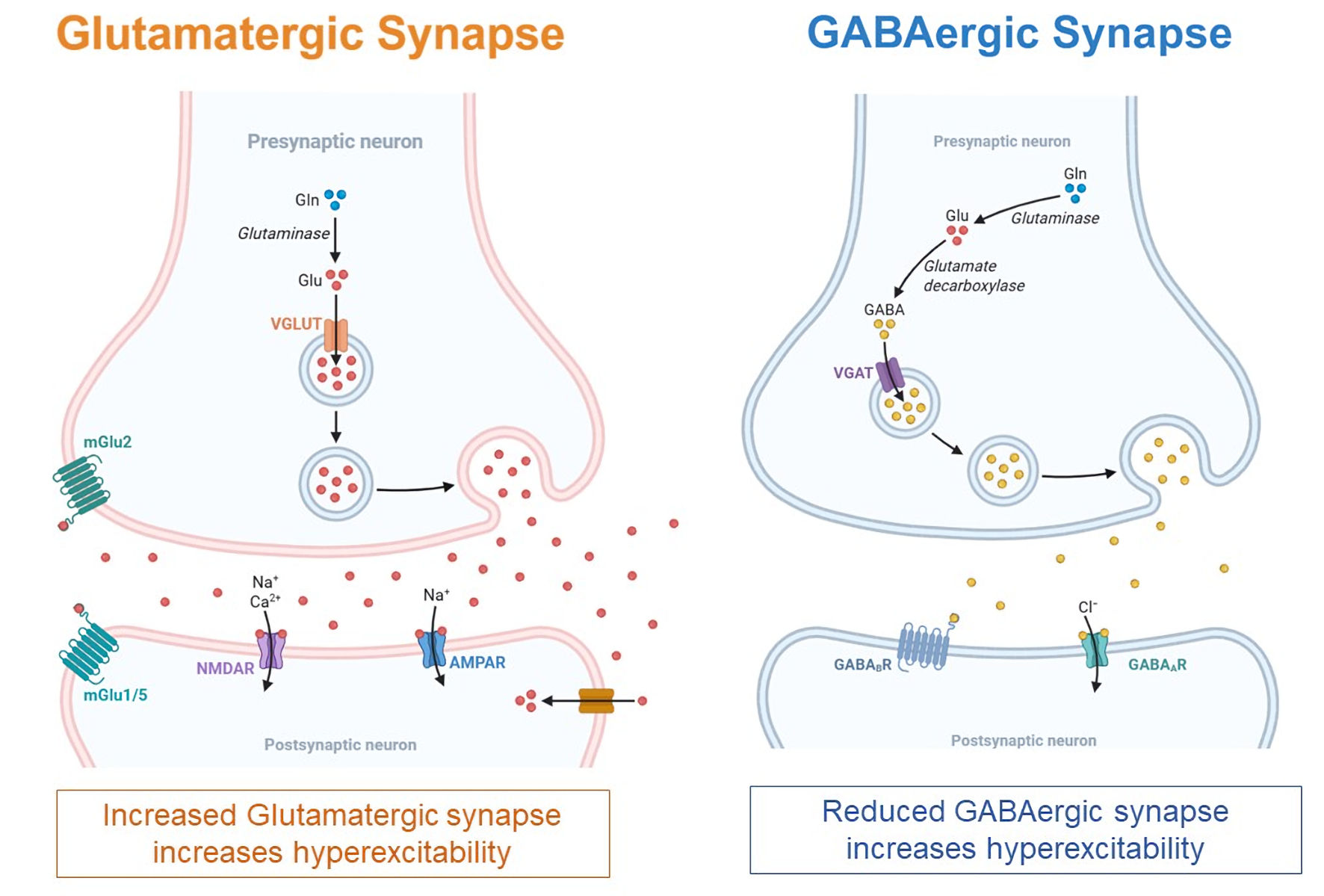
Tables
| Syndrome | Severity tier | DRE | Genetic associations | Typical age of onset | Estimated % of all epilepsy |
|---|---|---|---|---|---|
| ILAE: International League Against Epilepsy; DRE: drug-resistant epilepsy; TLE: temporal lobe epilepsy; ESES: electrical status epilepticus during sleep. | |||||
| Focal epilepsy type | |||||
| TLE (mesial & neocortical) | Severe | Yes | Often structural; LGI1 (lateral TLE), SCN1A, others | Adolescence to adulthood | 30-40% |
| Frontal lobe epilepsy | Moderate to severe | Varies | Often sporadic; DEPDC5 in familial cases | Childhood to adulthood | 15-20% |
| Parietal/occipital lobe epilepsies | Moderate | Varies | Mostly sporadic; some monogenic rare causes | Variable | 5-10% |
| Self-limited epilepsy with centrotemporal spikes (SeLECTS, rolandic) | Mild | No | Complex polygenic; rare familial cases with GRIN2A, DEPDC5 | Childhood (3 - 13 years) | 5-8% |
| Self-limited epilepsy with autonomic seizures (SeLEAS, Panayiotopoulos) | Mild | No | Unknown; sporadic; rare SCN1A variants reported | Childhood (1 - 14 years) | 2-5% |
| Childhood occipital visual epilepsy (Gastaut type) | Mild | No | Unknown/polygenic | Childhood (3 - 15 years) | 1-2% |
| Photosensitive occipital lobe epilepsy | Mild | No | CHD2, SCN1A, and other photosensitivity-related genes | Childhood/adolescence | 1-2% |
| Sleep-related hypermotor epilepsy (SHE) | Moderate | Varies | CHRNA4, KCNT1, DEPDC5, NPRL2, NPRL3, others | Childhood to adulthood | 1-2% |
| Familial TLE | Moderate | No | Autosomal dominant, complex genetics, e.g., LGI1 | Adolescence to adulthood | 1% |
| Focal epilepsy with structural/metabolic etiology | Severe | Yes | TSC1, TSC2, and other monogenic causes | Variable | 1% |
| Self-limited familial neonatal epilepsy (SFNE) | Mild | No | KCNQ2, KCNQ3 mutations | Neonatal (first days to weeks) | < 1% |
| Self-limited familial neonatal-infantile epilepsy (SFNIE) | Mild | No | SCN2A mutations | Neonatal to infancy | < 1% |
| Self-limited familial infantile epilepsy (SFIE) | Mild | No | PRRT2 mutations commonly implicated | Infancy (3 - 12 months) | < 1% |
| Generalized epilepsy type | |||||
| Juvenile myoclonic epilepsy | Moderate | Rarely | GABRA1, EFHC1, CACNA1H, others | Adolescence (12 - 18 years) | 5-10% |
| Childhood absence epilepsy | Mild | No | CACNA1H, GABRG2, SLC2A1 variants | Childhood (4 - 10 years) | 5-8% |
| Juvenile absence epilepsy | Mild to moderate | No | Polygenic; some voltage-gated channel genes | Childhood/adolescence (10 - 16 years) | 2-5% |
| Generalized tonic-clonic seizures alone | Moderate | Varies | Variable polygenic | Variable | 2-5% |
| Generalized epilepsy with febrile seizures plus (GEFS+) | Moderate | Varies | SCN1A, SCN1B, GABRG2 mutations | Infancy to childhood | 1-2% |
| Epilepsy with eyelid myoclonia (Jeavons) | Moderate | Varies | Polygenic, including CHRNA4 | Childhood to adolescence | 1% |
| Epilepsy with myoclonic absence | Moderate | Varies | Rare; limited data | Childhood/adolescence | < 1% |
| Combined epilepsy type | |||||
| Lennox-Gastaut syndrome | Severe | Yes | Multiple, including de novo variants in SCN1A, GABRA1, DNM1, CHD2 | Early childhood (1 - 8 years) | 3-5% |
| Dravet syndrome | Severe | Yes | SCN1A (about 80%), rare other genes (e.g., PCDH19, SCN2A) | Infancy (first year) | 1-2% |
| Epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS/ESES) | Severe | Yes | Variable; some cases with GRIN2A mutations | Childhood (3 - 10 years) | 1-2% |
| Epileptic spasms (West syndrome) | Severe | Yes | Diverse genetic causes: ARX, CDKL5, STXBP1, TSC1, TSC2, others | Infancy (3 - 12 months) | 1-2% |
| Epilepsy with myoclonic-atonic seizures (Doose syndrome) | Severe | Yes | SCN1A, SLC2A1, and others | Childhood (1 - 5 years) | 1% |
| Rasmussen syndrome | Severe | Yes | Autoimmune; genetic susceptibility unclear | Childhood (2 - 14 years) | < 1% |
| Febrile infection-related epilepsy syndrome (FIRES) | Severe | Yes | Possible immune-related genes (e.g., IL1R1); largely unknown | Children and young adults | < 1% |
| Ohtahara syndrome (early infantile epileptic encephalopathy) | Severe | Yes | Multiple including ARX, STXBP1, KCNQ2, SCN2A | Neonatal to early infancy | < 1% |
| Hemiconvulsion-hemiplegia-epilepsy (HHE syndrome) | Severe | Yes | Post-infectious, genetic susceptibility possible | Infancy and toddlerhood | < 1% |
| Unknown onset epilepsy type | |||||
| Epilepsy with unclassified seizures | Moderate | Varies | Unknown | Variable | 5-10% |
| Neonatal seizures of unknown onset | Severe | Yes | Various including KCNQ2, SCN2A mutations | Neonatal | 1-2% |
| Epileptic spasms of unknown origin | Severe | Yes | Diverse multiple genetic causes | Infancy | 1-2% |
| Unclassified neonatal/infantile epilepsy syndromes | Severe | Yes | Various genetic etiologies | Neonatal to infancy | < 1% |
| Intervention | Addresses emotions/behaviors | Includes family/school | Academic/learning focus | Psychotherapy techniques used | Best for |
|---|---|---|---|---|---|
| CBT: cognitive behavioral therapy. | |||||
| CBT | Yes | Occasionally | No | Yes | Discrete mood, emotional, behavioral, social issues |
| Psychoeducation | Some | Yes | Some | Indirect | Empowering families with epilepsy medical knowledge and engagement of resources |
| Educational therapy | Indirectly | Yes | Yes | Sometimes | Individualized academic intervention: attention/cognitive deficits |
| Psychoeducational therapy | Yes | Yes | Yes | Yes | Complex: overlapping psychoeducation, individualized psychotherapy and academic interventions |
| Condition | Associated epilepsy syndromes | Prevalence and effect on seizures |
|---|---|---|
| Prevalence figures represent estimates from heterogeneous studies. Direct causal relationships between comorbidities and seizure worsening require further empirical validation. DEE: developmental and epileptic encephalopathy; ESES: electrical status epilepticus during sleep. | ||
| Attention-deficit/hyperactivity disorder (ADHD) | Frontal lobe epilepsy, self-limited epilepsy with centrotemporal spikes (SeLECTS), childhood absence epilepsy, juvenile absence epilepsy, generalized tonic-clonic seizures alone, juvenile myoclonic epilepsy | 20-50% |
| Increases seizure frequency: associated with drug-resistant epilepsy and complex cases. More frequent generalized seizures (e.g., absence, tonic-clonic), and some focal seizures, especially frontal lobe epilepsy and self-limited epilepsy with centrotemporal spikes (SLECS) have higher ADHD comorbidity | ||
| Depression | Temporal lobe epilepsy (mesial & neocortical), juvenile myoclonic epilepsy, generalized tonic-clonic seizures alone, childhood absence epilepsy, juvenile absence epilepsy, Lennox-Gastaut syndrome, Dravet syndrome | 20-30% |
| Increases seizure frequency via stress, neuroinflammation: all seizure types, no specific type but generalized seizures often worsened by stress | ||
| Anxiety disorders | Temporal lobe epilepsy (mesial & neocortical), frontal lobe epilepsy, generalized tonic-clonic seizures alone, childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy | 20-25% |
| Triggers seizures, worsens control: generalized seizures and focal seizures sensitive to stress | ||
| Obsessive-compulsive disorder (OCD) | Temporal lobe epilepsy (mesial & neocortical), familial temporal lobe epilepsy | 10-14% (notably higher in temporal lobe epilepsy) |
| Associated with poorer seizure control, comorbid epilepsy: no specific seizure type identified; May worsen overall seizure burden | ||
| Autism spectrum disorder (ASD) | Lennox-Gastaut syndrome, Dravet syndrome, epileptic spasms (West syndrome), Ohtahara syndrome, epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS/ESES), epilepsy with myoclonic-atonic seizures (Doose syndrome) | 6-30% (higher in syndromic cases) |
| Increases epilepsy risk and seizure frequency, especially in syndromic and intellectual disability-associated cases: variety of seizure types seen: tonic-clonic, absence, complex partial seizures, higher rates in those with intellectual disability | ||
| Sleep disorders | Temporal lobe epilepsy (mesial & neocortical), sleep-related hypermotor epilepsy (SHE), generalized tonic-clonic seizures alone, childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy | 20-50% |
| Strong seizure trigger: focal seizures (especially temporal lobe), generalized tonic-clonic and absence seizures exacerbated by sleep deprivation | ||
| Intellectual disability | Lennox-Gastaut syndrome, Dravet syndrome, epileptic spasms (West syndrome), Ohtahara syndrome, epileptic encephalopathy with CSWS/ESES, epilepsy with myoclonic-atonic seizures (Doose syndrome), Rasmussen syndrome, febrile infection-related epilepsy syndrome (FIRES), hemiconvulsion-hemiplegia-epilepsy (HHE syndrome) | 10-40% (more in severe epilepsy/DEE) |
| Associated with severe epilepsy and higher seizure burden: higher likelihood of multiple seizure types including generalized and focal seizures | ||
| Learning disorders | SeLECTS, childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy, Lennox-Gastaut syndrome, Dravet syndrome, epileptic encephalopathy with CSWS/ESES | 20-50% |
| No direct influence on seizure types but worsens cognitive burden affecting management: no specific seizure types identified | ||
| Behavioral/conduct problems | Frontal lobe epilepsy, temporal lobe epilepsy (mesial & neocortical), generalized tonic-clonic seizures alone, juvenile myoclonic epilepsy, childhood absence epilepsy, juvenile absence epilepsy | 15-30% |
| Increase stress leading to poorer seizure control: stress-sensitive seizures including focal and generalized seizures | ||
| Migraine/headache disorders | Temporal lobe epilepsy (mesial & neocortical), generalized tonic-clonic seizures alone, juvenile myoclonic epilepsy | 8-15% |
| Associated with increased seizure frequency in migraine-epilepsy overlap: mainly generalized tonic-clonic seizures and complex partial seizures | ||
| Suicidality | Temporal lobe epilepsy (mesial & neocortical), juvenile myoclonic epilepsy, generalized tonic-clonic seizures alone, childhood absence epilepsy, juvenile absence epilepsy, Lennox-Gastaut syndrome, Dravet syndrome | 5-10% |
| Marker of severe psychiatric comorbidity associated with seizure control difficulty: indirect by association with depression/anxiety | ||