| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Review
Volume 000, Number 000, December 2024, pages 000-000

Molecular Signaling Pathways in Alzheimer’s Disease and Their Therapeutic Implications
Jahngeer Alama, c, Anushka Kalashb, Aishwarya Kulshresthab
aDepartment of Pharmacology, Faculty of Medicine, Aligarh Muslim University, Aligarh, Uttar Pradesh 202002, India
bDepartment of Pharmacology, School of Pharmaceutical Sciences, Jaipur National University, Jaipur, Rajasthan 302017, India
cCorresponding Author: Jahngeer Alam, Department of Pharmacology, Faculty of Medicine, Aligarh Muslim University, Aligarh, Uttar Pradesh 202002, India
Manuscript submitted October 29, 2024, accepted December 11, 2024, published online December 19, 2024
Short title: Signaling Pathways in AD Pathophysiology
doi: https://doi.org/10.14740/jnr869
| Abstract | ▴Top |
Alzheimer’s disease (AD) poses significant health challenges for the elderly due to its complex origins and heterogeneous etiology. Currently, there are no medications specifically aimed at halting its progression through any preventive signaling mechanism. Therefore, there is a pressing need to explore alternative pathways that could guide researchers towards new avenues in drug development for managing AD sustainability. This article is the first to simultaneously review several potential signaling pathways that were previously reported as individual signaling conduits in AD. Through meticulous analysis, we assess the pathophysiological roles of these pathways in AD. Extensive literature searches across databases including PubMed, Cochrane Library, Web of Science, Scopus, ResearchGate, Google Scholar, X-Mol, EBSCO, Loop, and Google, encompassing both research articles and highly cited reviews, were conducted. In addition to the cholinergic and N-methyl D-aspartate (NMDA) glutamate signaling pathways, we have observed four other less-explored, but promising molecular pathways relevant to AD onset and progression. We delved into the detailed signaling mechanisms of these pathways, spanning from preclinical to clinical studies. Remarkably, each pathway demonstrates distinct molecular signaling patterns and offers diverse perspectives on AD treatment. This review highlights six pivotal signaling pathways that may hold promise as future targeting pathways in managing AD.
Keywords: Alzheimer’s disease; Signaling pathway; Therapeutic target; Multi-target; Neuropharmacology
| Introduction | ▴Top |
Alzheimer’s disease (AD), initially described by German neuropathologist Alois Alzheimer in 1907, is characterized by neurotoxic presenile plaques. These plaques are believed to gradually impair memory and cognitive functions, leading to behavioral abnormalities in older individuals. Thus, AD manifests as difficulties in performing daily tasks, particularly those related to memory [1]. Increasing prevalence of AD is a growing societal concern, with the disease representing a significant global health crisis. From a statistical perspective, AD accounts for approximately 60% of all cases of dementia among individuals aged 65 years or older. Projections suggest that by the year 2050, the number of AD patients will reach 13.8 million [2, 3].
Despite extensive research, the precise etiopathophysiology of AD remains incompletely understood. However, two widely recognized pathological hallmarks are thought to play pivotal roles in its pathogenesis: the formation of extra-neuronal plaques composed of beta-amyloid (Aβ) peptides and the presence of intra-neuronal fibrillary tangles (NFTs) consisting of hyper-phosphorylated tau proteins. These patho-mechanisms are reported to contribute to neurodegeneration and further to onset and progression of AD [4, 5]. Numerous mechanisms have been proposed to elucidate the pathophysiological processes occurring in the brain during AD progression, ultimately culminating in symptomatic presentation. The elusive nature of AD pathologies poses challenges to identifying potential therapeutic targets, resulting in current pharmacotherapies primarily addressing symptomatic relief rather than disease prevention [6-8].
Contemporary pharmacological interventions for AD primarily target three distinct pathophysiological pathways: the cholinergic system signaling, N-methyl D-aspartate (NMDA) receptor signaling and antibody (Ab)-Aβ clearance. Currently, six pharmacological agents are available for AD management. Among these, three modulate cholinergic neurotransmission (Fig. 1): rivastigmine (Exelon), galantamine (Razadyne), and donepezil (Aricept), while one acts as an NMDA receptor antagonist (Table 1) [9-14], namely memantine (Namenda). Additionally, two recently approved therapeutic agents, aducanumab (Aduhelm) and lecanemab (Leqembi), are antibodies designed to reduce neuronal levels of neurotoxic Aβ-oligomers or plaques [15, 16]. Despite the limited availability of medications for AD treatment, existing therapy options have demonstrated notable side effects upon prolonged use [17]. This challenging scenario underscores the necessity to explore alternative neurobiological pathways relevant to AD pathophysiology. Moreover, beyond these approved classes of anti-AD compounds targeting neurotransmission of acetylcholine (ACh) and glutamate, researchers are continued to explore other chemical entities targeting diverse therapeutic pathways, including the signaling pathways involving neurotransmitters other than ACh and glutamate (Table 2) [18-23]. Numerous studies have highlighted the potential therapeutic value of various novel pathological pathways in mitigating AD, with investigations conducted through in-silico, in-vitro, in-vivo or preclinical, and clinical testing [24, 25]. The objective of this study was to investigate essential signaling pathways that could serve as potential targets for the management of AD.
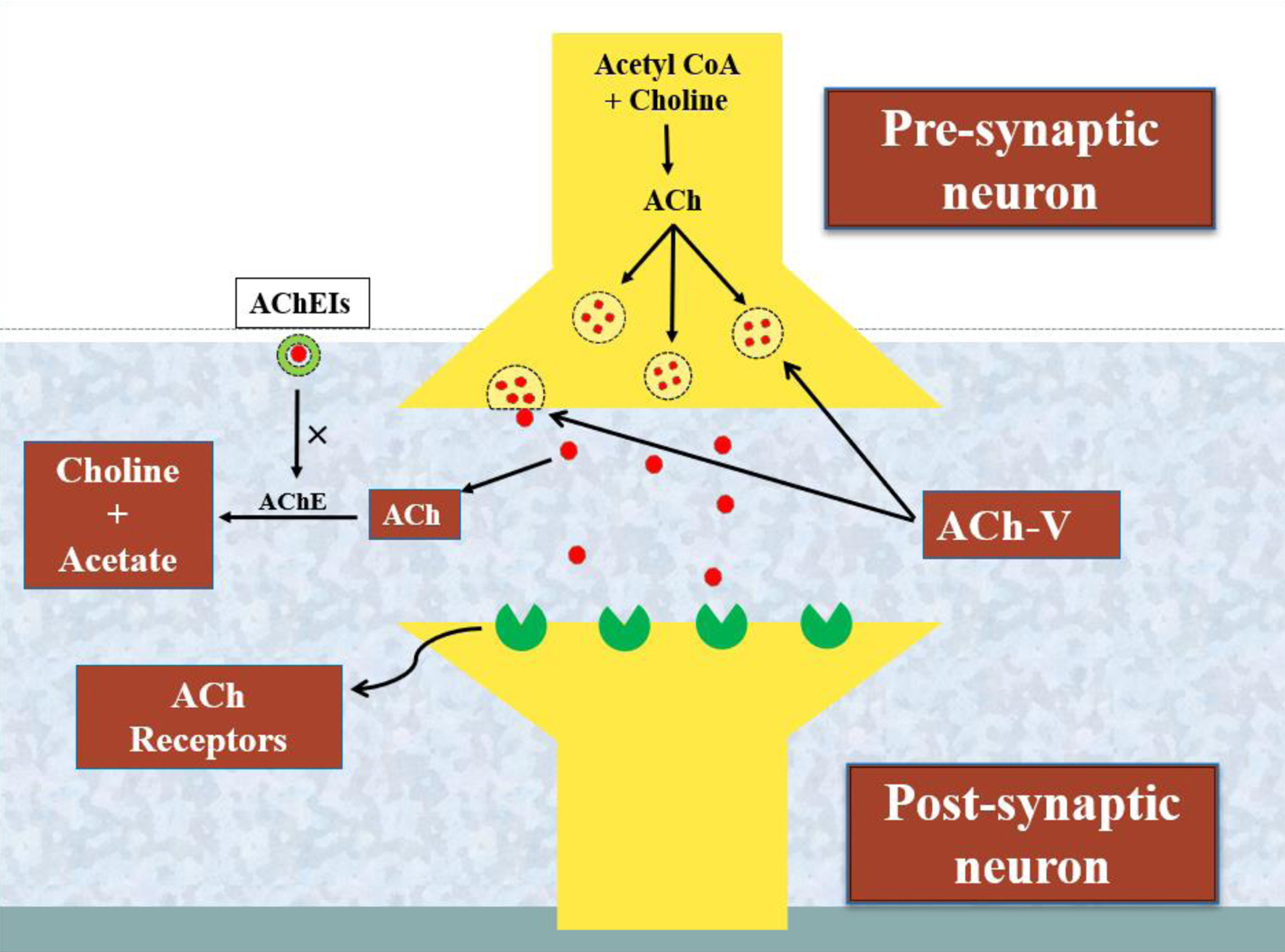 Click for large image | Figure 1. A schematic representation of the activity of acetylcholinesterase and the processing of the neurotransmitter acetylcholine. The enzyme choline acetyltransferase converts acetyl-CoA and choline into acetylcholine (ACh). Furthermore, ACh is released from ACh vesicles (ACh-V) to bind ACh receptors and performs a variety of neurological tasks that are impaired by the degrading action of another enzyme, acetylcholinesterase (AChE). In this way, the acetylcholinesterase inhibitors (AChEIs) reverse the brain functioning by blocking the activity of AChE in Alzheimer’s disease (AD). |
 Click to view | Table 1. Important Signaling Cascades and Their Primary Targets in the Pathophysiology of AD [9-14] |
Click to view | Table 2. Drug Entities Under Phase 3 Clinical Trials Against Possible Molecular Cascades for the Potential Futuristic Therapeutics in AD Treatment Scenario |
| Literature Review | ▴Top |
To ensure a comprehensive review, we conducted a thorough search of major scholarly databases, including PubMed, Cochrane Library, Web of Science, Scopus, ResearchGate, Google Scholar, X-Mol, EBSCO, Loop, and Google. Our search utilized keywords related to AD pathophysiology, novel signaling pathways, multi-target therapies, neurotransmission disruptions, and clinical trials. This extensive search yielded a diverse selection of original as well as review articles, which forms the foundation of our analysis.
Following the literature review, the authors of this manuscript have identified numerous potential targets of a distinguished substance known to modulate neuronal dysfunctions, specifically in the progression of the disease. Besides the cholinergic system and NMDA receptor signaling pathways, four additional neurotransmission signaling pathways are discussed in the context of their potential therapeutic roles in AD: serotonin receptor signaling, GABAergic system signaling, adenosine receptor (AR) signaling, and histaminergic system signaling. These pathways hold promise as potential drug targets for AD management that could shape the future of AD therapeutics (Table 1).
Emerging signaling pathways in AD pathophysiology
In this study, we have comprehensively examined the physiology of neuromodulation within the pathological pathways implicated in AD, whether directly or indirectly. Our focus lies on analyzing insights gleaned from both preclinical and clinical investigations (Table 2) concerning these signaling pathways. This article emphasizes the significance of pathway-specific pathological consideration, which is poised to guide researchers towards identifying pivotal signaling targets essential for developing potential pharmacological interventions for AD management. The detailed discussion of these crucial signaling pathways underscores their importance in advancing our understanding of AD.
Serotonin receptor signaling
The literature extensively documents serotonin (5-hydroxytryptamine (5-HT)) as a pivotal neurochemical investigated across brain and gastrointestinal contexts due to its multifaceted regulatory functions, including mood control, aggression modulation, sleep regulation, pain perception, body temperature maintenance, and appetite regulation [9]. Serotonin is synthesized and released by serotonergic neurons clustered in the raphe nuclei of the brain stem’s pons. Its physiological roles encompassing neural development and differentiation underscore its significance in sustaining learning processes and memory storage throughout adulthood and aging (Fig. 2). Dysregulated serotonin metabolism or impaired serotonergic neuron function has been implicated as an etiological factor in various central nervous system (CNS) disorders, including AD (Table 1). Reduced levels of serotonergic neurons are associated with symptomatic manifestations of AD, although the extent to which serotonergic neuron atrophy contributes to AD pathogenesis remains unclear [9]. Studies utilizing an amyloid precursor protein (APP)-transgenic mouse model have reported Aβ deposits at serotonergic neuron projection sites, suggesting a potential link between Aβ protein toxicity and serotonergic neuron degeneration. Furthermore, diminished serotonin levels in projection areas [10] may also contribute to AD progression.
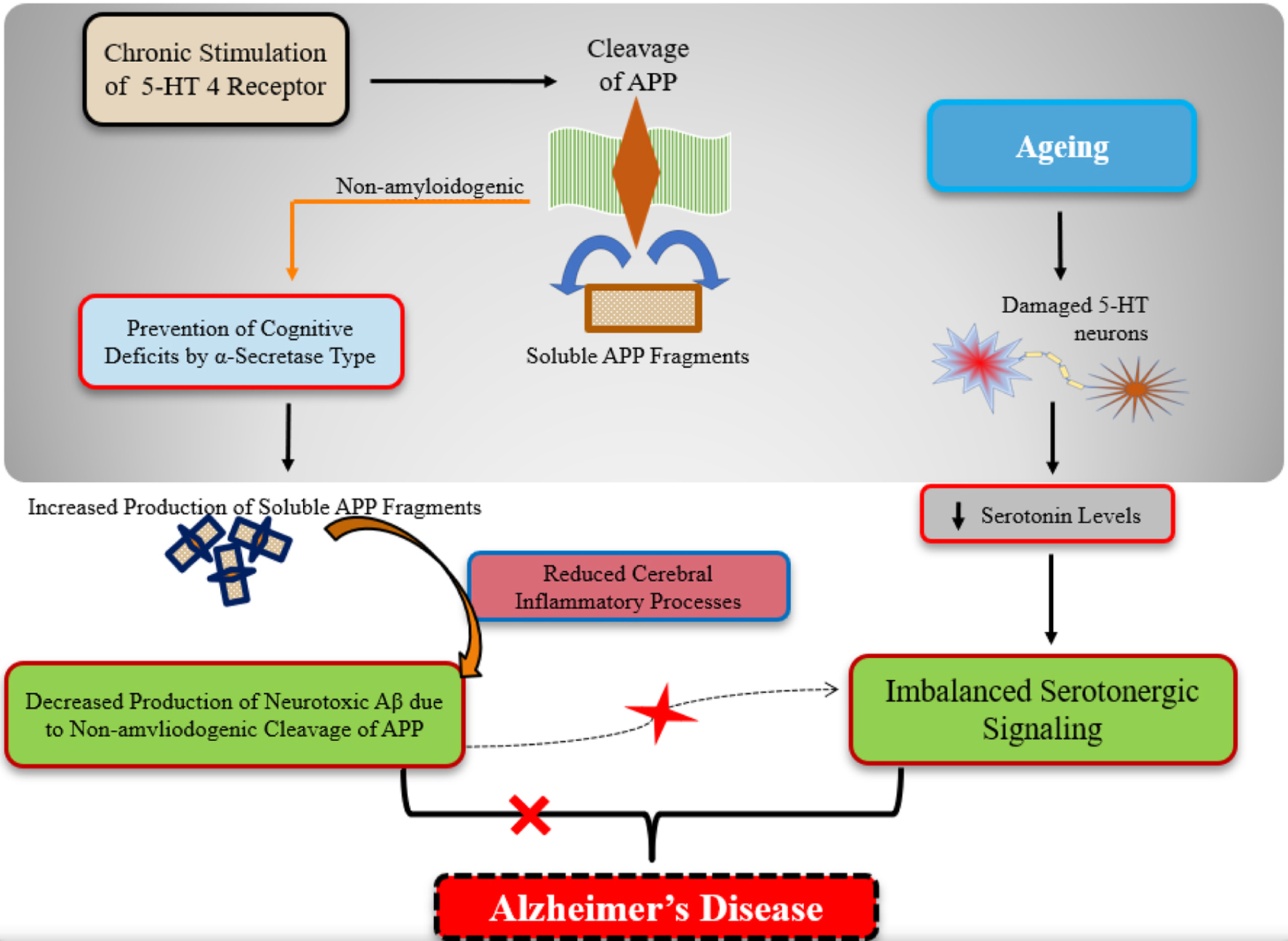 Click for large image | Figure 2. A diagrammatic presentation of the role of the serotonergic system signaling in aging and Alzheimer’s disease (AD). As we grow older, the serotonergic neurons tend to get damaged which leads to impair the normal serotonergic signaling in elderly. In case of AD, the chronic stimulation of the serotonin type-4 receptor leads to the non-amyloidogenic α-secretase cleavage of amyloid precursor proteins (APPs). This cleavage type results in the increased production of non-neurotoxic beta-amyloid (Aβ) fragments (i.e., soluble forms) and the depleted production of neurotoxic Aβ (i.e., non-soluble forms). |
The activation of 5-HT6 receptors (5-HT6Rs) has been shown to directly interact with and activate the enzyme cyclin-dependent kinase-5 (CDK-5), a key tau phosphorylating kinase, while also modulating non-proline directed protein kinase activity. However, the precise impact of 5-HT6R-mediated CDK-5 activation on tau pathophysiology remains unclear [25, 26], necessitating further investigation to elucidate its role in the production of NFTs. Recent findings from the clinical trials phase-II involving a combination of donepezil (an ACh esterase (AChE) enzyme inhibitor) and idalopirdine (a 5-HT6R antagonist) and phase-III (Table 2) have demonstrated improved cognition in patients with moderate AD. At the cellular level, inhibition of 5-HT6R has been shown to prevent further mammalian target of rapamycin (mTOR) signaling rather than activating neuromodulation mediated by the stimulatory GDP-directed (sG) proteins (sGP).
Serotonin operates through specific 5-HT receptors (5-HTRs), which are categorized into seven receptor classes based on their structural and functional characteristics. All 5-HTRs are seven-transmembrane protein subunits that interact with intracellular regulatory proteins via heterotrimeric G-proteins, except for the transmitter-gated Na+/K+ channel and 5-HT3R. The 5-HT1R inhibits adenylyl cyclase (AC) activity through coupling with inhibitory G-proteins (iGP), while the three isoforms of 5-HT2R (5-HT2A, 5-HT2B, and 5-HT2C) activate phospholipase-C (PLC) by coupling with heterotrimeric subtype alpha11 G-proteins (qGP). The effector system for 5-HT5AR and 5-HT5BR remains unclear, although evidence suggests activation of ACs through coupling with PTX-sensitive iGP. Additionally, specific coupling between 5-HTR subfamilies (5-HT4R, 5-HT6R, and 5-HT7R) and sGP can activate AC [26, 27]. It has been hypothesized that protein phosphatase 2A (PP2A) inhibitors selectively upregulate serotonin transporter (SERT) phosphorylation, suggesting PP2A may target phosphorylation sites on SERT itself. Furthermore, activation of 5-HT4 and 5-HT7 receptors can stimulate G12/13 proteins [28], leading to morphological changes at the cellular level. This discussion represents that it is still a matter of debate whether actually the inhibition or activation of 5-HTRs would be beneficial clinically. Therefore, the serotonin receptor signaling has been a prominent neuromodulation pathway in the brain to study, with its diverse signaling targets as discussed above, which should be investigated for the development of novel drug agents for AD treatment.
GABAergic system signaling
Gamma-amino butyric acid (GABA), the principal inhibitory neurotransmitter, plays a pivotal role in modulating excitability and responsiveness in cortical regions, as well as in synchronizing neuronal signaling within the cortex. The widespread regulation of GABA neurotransmission in the brain contributes significantly to various biochemical processes in the body. Both the stimulation and inhibition of GABA receptors (rGABA) have been shown to have beneficial effects in AD. Therefore, the use of GABAergic compounds can influence learning and memory processes and performance. In this context, researchers have investigated the potential role of rGABA in streptozotocin-induced behavioral and biochemical abnormalities in rats [29].
Numerous lines of evidence support the pathological involvement of the GABAergic system in the neurodegenerative processes underlying AD. Previous research has presented a complex, intricate, and often conflicting portrayal of AD-associated GABA remodeling. Alterations in GABA receptor subtypes (GABA-a, -b, and -c) may represent key factors in the pathogenesis of both early-onset AD (EOAD) and late-onset AD (LOAD). GABA-a/b receptors are ligand-gated chloride channels, while GABA-b receptors are G-protein coupled receptors. In comparison to the well-documented deficiencies in the excitatory cholinergic and glutamatergic systems in AD, findings related to the GABAergic system in the nervous system have been notably less consistent [30]. The dynamic balance between inhibitory GABA and excitatory glutamate neurotransmitters is crucial for proper neuronal function. Dysregulated synaptic activity is implicated in various neurological disorders, including AD-type dementia, where neuronal hyperexcitability is considered a major contributor to neuronal cell death [31]. Consequently, enhancing GABAergic neurotransmission with GABA receptor agonists (Table 1) might be inverse to the NMDA pathophysiology of AD and may represent a promising approach to mitigate glutamate-hyperexcitability and prevent neuronal cell death in AD in the future.
Adenosine receptor signaling
1) Adenosine type-1 (A1) receptors in AD pathophysiology
Adenosine exerts its inhibitory effects on synaptic communication through the modulation of neurotransmitter transmission and release via A1 receptor activity inhibition. Specifically within the hippocampus, adenosine inhibits ACh release. This modulation of synaptic activity by adenosine is implicated in various learning and memory tasks (Fig. 3), suggesting a role for synaptic plasticity in cognitive processes across different brain regions [32]. Activation of ARs disrupts learning and memory tasks in animal models, while non-selective blockade with theophylline or caffeine (Table 1), or selective inhibition of A1 and A2a receptors, enhances performance in behavioral tasks [33].
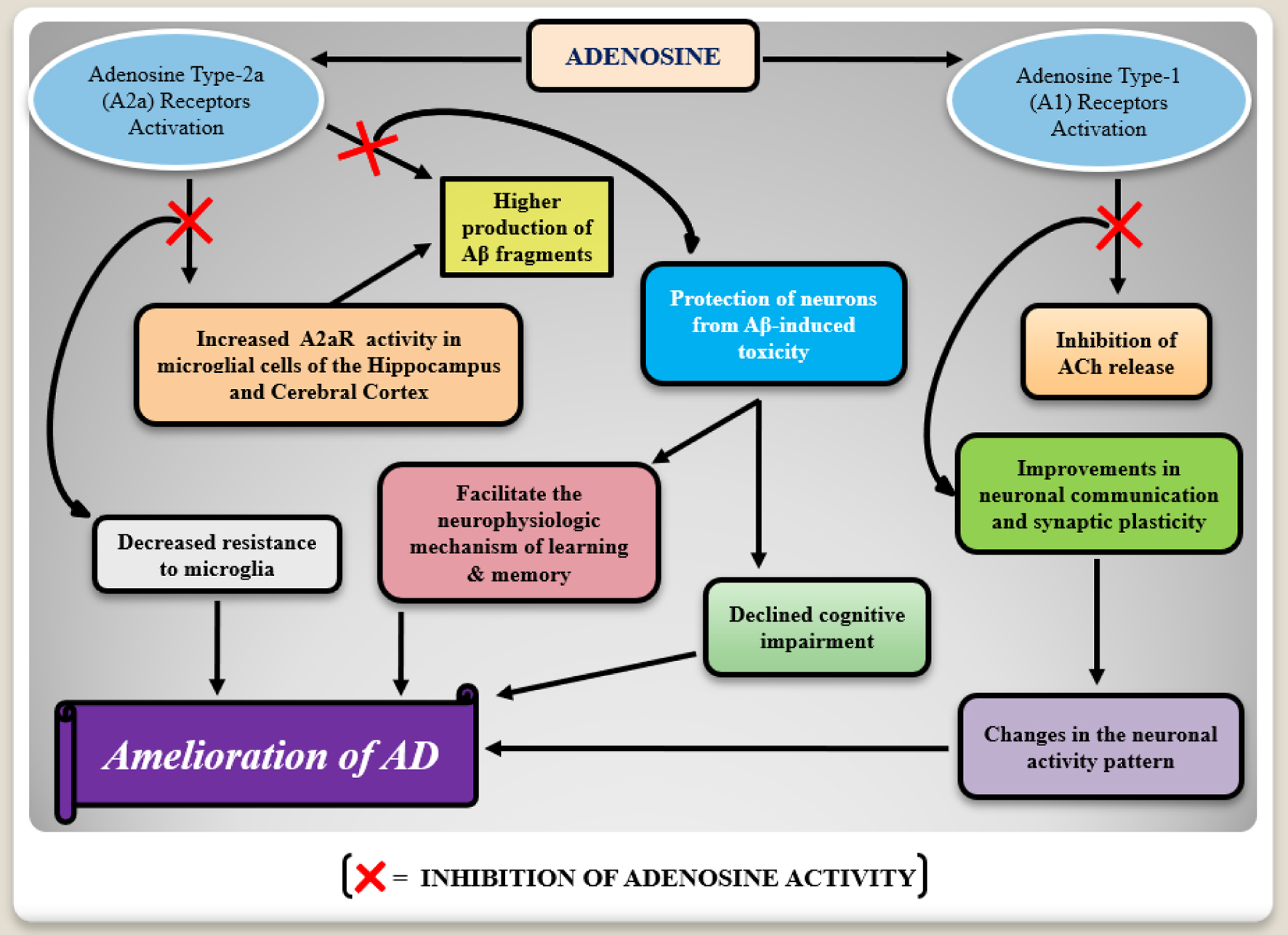 Click for large image | Figure 3. A flow chart mechanism of the adenosine type-1 (A1) and type-2 (A2) receptors involvement in Alzheimer’s disease (AD) pathophysiology. Adenosine receptors activation seems to induce degenerative consequences. Increased expressions of type-2 adenosine receptors (A2aR) in microglia may favor the production of neurotoxic beta-amyloid (Aβ) proteins, whereas type-1 adenosine receptors are responsible for acetylcholine (ACh) release inhibition which leads to the degenerative changes in the neuronal activity pattern in AD. In this way, the inhibition of these receptors activity presents the cognition improvement. |
A1 receptors are notably abundant in the cornu ammonis-1 (CA1) region of the hippocampus in healthy brains, but their expression is reduced in the hippocampus and basal ganglia of aging and AD brains. Altered A1 receptor expression in AD patients is associated with decreased adenosine agonist and antagonist bindings in regions like the dentate gyrus (DG), CA1, and CA3 [34]. The precise role of A1 receptors in AD pathology remains unclear, although evidence suggests involvement in abnormal APP processing and formation of NFTs. Hence, we must investigate into this role of adenosine type-1 receptors and their signaling in a more transparent fashion for managing AD-associated neurodegenerative consequences connected with this hypothesis.
2) Adenosine type-2a (A2a) receptors in AD pathophysiology
A2a receptors exhibit lower expression levels in healthy brains, although this expression pattern may change in pathological conditions. Limited data are available regarding the distribution of A2a receptors in AD patients, but increased expression has been observed in specific brain regions such as the hippocampus and cerebral cortex, particularly in microglia [33, 35]. In addition to AD, elevated A2a receptor expression has been noted in patients with Parkinson’s disease, stress conditions, and Pick’s disease. Modulation of A2a receptors is implicated in neurophysiological mechanisms underlying learning and memory, offering potential relief from cognitive impairment in AD patients following combating their expression levels [35, 36].
Brain-derived neurotrophic factor (BDNF), a receptor for specific amino alkanoic acids belonging to the neurotrophin family (i.e., tropomyosin receptor kinase B, TrKB), is highly expressed in the hippocampus. Blocking A2a receptors and depleting extracellular adenosine abolishes BDNF’s facilitated effect on long-term potentiation (LTP), a process crucial for synaptic plasticity underlying memory formation. Neurotrophins serve as potent survival factors for both developing and mature neurons. Activation of TrKB through transactivation pathways, such as using A2a receptor agonists, can stimulate neuronal survival. However, clinical efficacy of A2a receptor agonists is limited due to excitotoxic glutamate-mediated neuronal damage, leading to desensitization of A2a receptors even after short exposure to agonists [32, 35, 36]. Although the in-vitro stimulatory effects of A2a receptors are promising, their translation to the in-vivo setting has limitations. Nevertheless, modulating A2a receptor activity could represent a neuroprotective strategy in the treatment of AD (Fig. 3).
3) Relationship among caffeine, ARs, and AD
It is noteworthy that CNS activation by caffeine leads to increased levels and activity of ACh in the brain. Caffeine, a prominent component in beverages and natural products, acts as an antioxidant, protecting neurons from oxidative damage by inhibiting free radical formation and reducing the risk of neurodegeneration (Table 2). Additionally, caffeine enhances cerebral glucose utilization rates, thereby contributing to improved cognitive function. As a neuromodulator, caffeine has associative effects on motor behavior, information processing, and cognitive functioning. Acting as a non-selective antagonist of A1 and A2a receptors, caffeine inhibits pre-synaptic glutamate release via A1 receptor activation, thereby dampening excitatory synaptic transmission [11]. Furthermore, post-synaptically, A1 receptor activation inhibits potassium conductance, ultimately leading to neuronal hyperpolarization. A2a receptors have been implicated in regulating synaptic transmission and LTP in the CA1 region of the hippocampus, suggesting potential relevance to the pathogenesis of AD [32]. Despite limited studies examining the relationship between coffee consumption and cognitive decline, dementia, or AD [33], it is proposed that further investigation into the specific pathways through which caffeine exerts its beneficial effects via AR-mediated signaling in AD pathophysiology could enhance our understanding of caffeine’s role in ameliorating neurodegeneration-associated brain complications such as memory impairment and cognitive dysfunction.
In the nutshell, we conclude that adenosine signaling more potential found with the concept of its antagonism rather than activation. Therefore, we recommend researcher to perform detailed studies for looking into new drug development strategies by targeting AR signaling.
Histaminergic system signaling
Histamine, a biogenic monoamine, acts as an inflammatory mediator released primarily from mast cells, neurons, and basophils in response to various external and internal stimuli, including the processing of the antibody immunoglobulin E release. Its presence in the bloodstream triggers the activation of various immunological cells, including natural killer cells, epithelial cells, and T and B lymphocytes. Histamine is recognized for its pivotal regulatory role in the autoimmune model of multiple sclerosis, known as experimental allergic encephalomyelitis [37]. In addition to its immunological effects, histamine facilitates cell growth, tissue regeneration, wound healing, and other physiological processes. Moreover, it modulates the T-helper cells type-1 and 2 balance and hematopoiesis. Histamine (H) is categorized into four subtypes, namely H1, H2, H3, and H4. G-protein coupled receptors are present in all four histamine receptors. Human H3 receptors (H3R) share 40% homology with human H4 receptors (H4R) [12].
Histamine enhances the synthesis and release of various proinflammatory mediators, including chemokines such as chemokine (CC motif) ligand-5 (CCL-5 or RANTES) and cytokines such as interleukin (IL): IL-1, 3, 6, and 8, leading to the development of allergic-inflammatory responses across different cell types and tissues. While it remains uncertain whether abnormalities in brain histamine processing significantly contribute to neurological disorders [37], the development of agonists and antagonists targeting histamine receptors is considered crucial for the treatment of brain-related illnesses (Table 1). Particularly, the H3R has emerged as a promising target for conditions like narcolepsy, neuropathic pain, and cognitive impairment. Many cellular behaviors induced by histamine in leukocytes, including cytokine production and cell migration, are also mediated by the H4R. Recent findings suggest that histamine signaling via H4R exhibits anti-pruritic and anti-nociceptive properties. Additionally, histamine may influence the function of the brain’s striatum by regulating thalamo-striatal synapse dynamics, ultimately facilitating thalamic input [12, 37]. Despite these insights, the precise role of histamine in regulating brain activities in AD remains incompletely understood, both from preclinical and clinical perspectives.
Cholinergic system signaling
Cholinergic neurotransmission is primarily regulated by the neurotransmitter ACh. In AD, there is typically a decrease in ACh levels in the brain, which correlates with symptoms related to memory and cognition dysfunction. The enzyme AChE plays a crucial role in breaking down ACh to maintain brain homeostasis under normal conditions; however, this enzymatic activity is considered pathological in case of AD (Table 1). Thus, inhibiting AChE activity is seen as a potential therapeutic approach for AD treatment, as evidenced by the use of the United States Food and Drug Administration (US FDA)-approved anti-cholinergic drugs (Fig. 1).
While it was initially hypothesized that AChE inhibition might also help counter the formation of presenile plaques, conclusive evidence for these remains elusive [38]. Studies have shown that as AD progresses, there is significantly reduced AChE activity in the temporal lobe and hippocampus [16, 39]. This understanding led to the development of the first AChE inhibitor, tacrine, approved for mild to moderate AD in 1993 but later withdrawn from the market in 2013 due to severe side effects. Currently, novel approaches such as bio-oxidizable prodrugs are being explored to improve AChE inhibition in AD treatment.
Additionally, butyrylcholinesterase (BuChE), a similar enzyme to AChE, has also been implicated in AD pathogenesis, leading to investigations into its inhibition as a therapeutic target. Numerous compounds targeting either AChE or BuChE have undergone preclinical or clinical testing, with some still in various phases of clinical trials [7]. In addition to targeting individual enzymes, there is interest in exploring multi-target strategies with new chemical entities to potentially reduce side effects in AD management (Table 2). Overall, AChE and BuChE enzymes represent valid and promising targets for therapeutic intervention in AD [40].
NMDA-glutamate receptor signaling
Cholinergic system-based therapy, while improving cognitive abilities, lacks neuroprotective properties and was the sole medication available for AD until recently. Numerous prospective neuroprotective medications evaluated in clinical trials were unsuccessful due to poor tolerability. However, memantine emerged as a new drug, the first approved by both the US and European Union for AD treatment. Memantine acts as an antagonist by blocking NMDA receptors, which mediate excitotoxicity following glutamate binding (Table 1). Excitotoxicity refers to excessive stimulation of these receptors by glutamate (Fig. 4), leading to neuronal damage or death [41]. Calcium ion influx through receptor-associated ion channels results in free radical generation, contributing to excitotoxic neuronal damage via NMDA receptor over-activation [13]. Neuroprotective drugs that completely inhibit NMDA receptor activation often entail undesirable side effects.
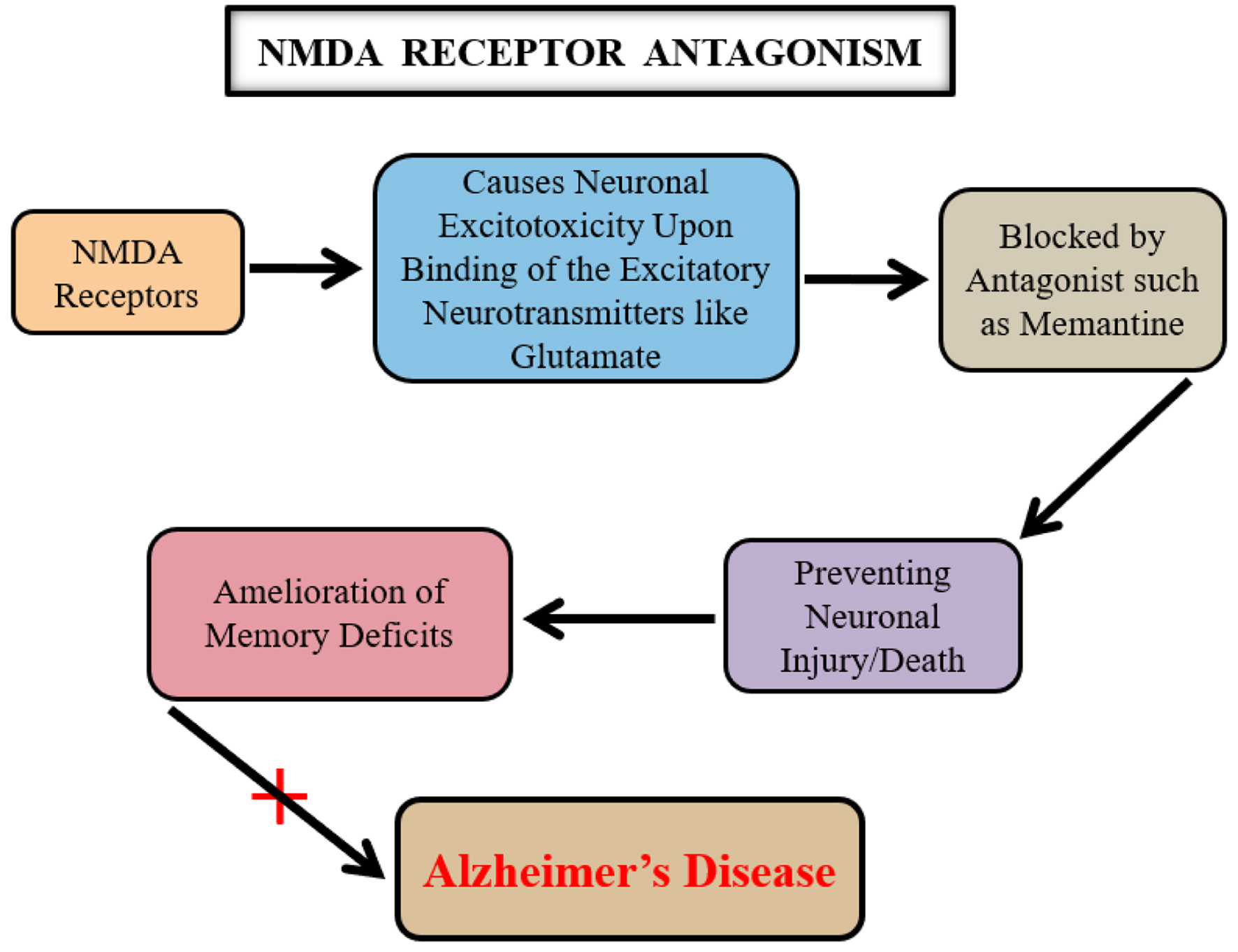 Click for large image | Figure 4. A key relationship among the N-methyl D-aspartate (NMDA) receptor signaling, glutamate, and Alzheimer’s disease (AD). The neurotransmitter glutamate causes neuronal excito-toxicity upon its binding to the NMDA receptors. A prolonged activation of the NMDA glutamate receptors leads to the neuronal dysfunctioning which is potentially inhibited by using an NMDA receptor antagonist such as memantine. This mechanistic effect of memantine is therefore counted as beneficial in AD. |
Memantine selectively blocks excessive NMDA receptor activity without disrupting normal receptor function, thereby clinically benefiting cognition and memory deficits by normalizing the glutamatergic system. Second generation memantine derivatives and other NMDA receptor antagonists are currently undergoing various phases of drug development [42], with researchers aiming for agents with enhanced neuroprotective properties (Table 2). NMDA receptor signaling remains a pharmacologically valuable target in AD management.
Therapeutic approaches for AD management
Acetylcholinesterase inhibition
The decline in cholinergic transmission observed in AD has led to the development of AChE inhibitors as the primary line of treatment. These agents offer clinical benefits including improvements, stabilization, or partial decline in cognition, memory, and behavioral abnormalities. The common mechanism of action among this class of agents involves increasing available ACh levels by inhibiting the catabolic enzyme AChE. There is substantial evidence indicating that various AChE inhibitors, including the US FDA-approved drugs such as donepezil, galantamine, and rivastigmine, reduce AChE activity by impeding the breakdown of neurotransmitter ACh within several brain regions, both preclinically and in patients with AD. Furthermore, a significant correlation exists between AChE inhibition and observed cognitive enhancement (Table 1). Therefore, AChE inhibition remains the primary therapeutic target for managing AD patients [43, 44]. In this context, there is a rationale for exploring more potent anti-AChE agents and cholinergic receptor modulators (Table 2) than the currently available cholinergic therapeutics, which have been associated with undesired effects in a substantial portion of AD patients during long-term management.
NMDA-glutamate receptor antagonism
In the treatment of mild to severe AD, NMDA receptor antagonism is a viable approach supported by the US FDA approval of memantine, an NMDA receptor antagonist. Memantine functions by reducing excitotoxicity induced by the glutamate neurotransmitter, thereby mitigating neurodegeneration resulting from excessive glutamatergic neurotransmission (Fig. 4). Additionally, memantine has demonstrated efficacy in reducing tau phosphorylation levels and protecting neurons from Aβ-induced neurotoxicity. Thus, the pharmacological potential of memantine lies in its role as an NMDA receptor antagonist [45, 46]. This discussion suggests the possibility of identifying novel NMDA receptor antagonists with minimal side effects and greater potency than memantine (Table 2).
Serotonin receptors modulation
The 5-HT6R, one of the latest cloned serotonin receptors, is intricately linked to AC via a soluble sGP, making it a well-known member of the 5-HTR family. Among various brain regions, including the corpus striatum, hippocampus, and cortex, this receptor exhibits particularly robust expression within the CNS. Notably, antipsychotic medications such as loxapine and clozapine (Table 1), as well as tricyclic antidepressants like amitriptyline, clomipramine, and amoxapine, have demonstrated strong affinity for the 5-HT6R. Given their prevalence in the limbic region of the brain, which plays a crucial role in mood regulation, these receptor proteins have garnered considerable attention. Significantly, a notable reduction in the density of 5-HT6Rs has been observed in cortical brain regions of patients with AD [14]. This insight into serotonin receptor activity underscores the importance of these receptors in brain function. Consequently, further research, particularly focused on elucidating the pathological processes of AD, holds great significance and has the potential to facilitate the development of new drugs (Table 2) targeting novel therapeutic pathways, such as the modulation of 5-HTR expression.
GABA neurotransmission upregulation
GABA is a physiologically inhibitory neurotransmitter within the nervous system, exerting a suppressive effect on neural activity. GABAergic neurons densely innervate cholinergic and glutamatergic neurons. Dysfunction within the GABAergic system has been implicated in impairments of psychological function in humans. Severe cases of AD have been associated with significant reductions in GABA levels, which may contribute to the behavioral and psychological symptoms of the disease [47]. Correction of hyper-excitability and prevention of cell death in AD could potentially be achieved through the use of GABA agonists such as homotaurine to enhance GABAergic neurotransmission (Table 1). Therefore, exploring upregulators of GABA neurotransmission represents a novel and promising approach for addressing AD-related behavioral and psychological symptoms.
Adenosine receptors antagonism
Adenosine and ARs have emerged as significant therapeutic targets due to their dual roles within the CNS, which involves regulating cognition under normal conditions and modulating neural activity during diseased states. Adenosine exerts its effects through both inhibitory and facilitatory actions on A1 and A2A receptors, respectively, thereby integrating signaling pathways involving neurochemicals such as glutamate, dopamine, and BDNF to modulate synaptic plasticity, specifically in the form of long-term depression-LTP. This molecular and cellular modulation of ARs contributes to the regulation of cognitive functions [38, 48, 49]. Targeting ARs for drug development in AD management represents a crucial strategy (Table 2), particularly in the absence of preventive treatments despite a century of research into the disease’s origins. Therefore, the drugs such as caffeine and istradefylline, which act as antagonists of AR, are being explored to enhance brain function, particularly in mental impairment states observed in Parkinson’s disease and AD patients.
Histaminergic system modulation
The actions of histamine are mediated through the four G-protein-coupled aminoalkane receptors (H1R-H4R) located on its surface, thereby implicating the histaminergic system in the treatment of brain disorders [13]. Specifically, H1R and H4R molecules are considered pivotal in allergic inflammation processes. Recent studies have indicated that antagonism of H3R [42] and modulation of H2R may present alternative therapeutic approaches in the management of AD [50]. The significance of histaminergic receptor signaling suggests that down-regulating histamine activity and modulating its receptors could serve as effective therapeutic avenues in AD pathophysiology [51]. However, the precise role of histamine remains a subject of debate. Some researchers have noted that benadryl, an antihistamine with inverse agonist activity on H1R, may elevate the disease risk, highlighting antihistamines’ potential role as anticholinergics.
| Conclusion and Future Directions | ▴Top |
The discussion presented in the article underscores the continued relevance of AChE as a promising therapeutic target for AD management, given the prevalent use of this signaling pathway in numerous medications and ongoing clinical trials aimed at identifying more potent anti-AD compounds. Additionally, targeting NMDA receptor signaling remains a valid approach in AD treatment. Researchers have also highlighted the detrimental effects of disrupted serotonin metabolism and reduced serotonergic neuron activity as potential etiological factors in AD, suggesting that addressing these factors may constitute an important additional therapeutic target for addressing neuronal metabolic dysfunction in AD. Enhancing GABAergic neurotransmission through the use of rGABA agonists, such as homotaurine, shows promise in mitigating neuronal death by correcting hyperexcitability. AR antagonists like caffeine have demonstrated effective neuromodulatory actions in AD patients. However, the role of histamine in modulating brain activity, whether through receptor inhibition or activation, remains poorly understood in AD pathophysiology and requires further detailed investigation as a potential target within the histaminergic system for anti-AD treatment. Overall, the article extrapolates and elucidates the specific molecular underpinnings of these pathways based on their preclinical and clinical studies. Presently, no single pathway stands out as paramount for targeting, as each neuromodulatory pathway represents a crucial target in the drug development landscape. Consequently, the authors advocate for the design and development of multi-target agents rather than solely focusing on single-target approaches in drug discovery endeavors.
Acknowledgments
The authors are thankful to all the affiliated institutions for providing the internet and library facilities to carry out the literature regarding the present work.
Financial Disclosure
The authors did not receive support from any organization for the submitted work.
Conflict of Interest
None to declare.
Authors Contributions
Conceptualization: Jahngeer Alam; literature search: Jahngeer Alam, Anushka Kalash, and Aishwarya Kulshrestha; drafting: Anushka Kalash and Aishwarya Kulshrestha; review, editing, and refining: Jahngeer Alam; supervision: Jahngeer Alam.
Data Availability
This review article contains no datasets generated or analyzed during the current study.
| References | ▴Top |
- Gomez-Gallego M, Gomez-Gallego JC. Predictors of caregiver burden of patients with Alzheimer disease attending day-care centres. int j environ res public health. 2021;18(20):10707.
doi pubmed - Alam J, Kalash A, Hassan MI, Rahman SZ. Agents at the peak of US FDA approval for the treatment of Alzheimer's disease. Neurol Res. 2024;46(4):318-325.
doi pubmed - 2023 Alzheimer’s disease facts and figures. Alzheimer's Dement. 2023;19:1598-1695.
doi - Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. 2022;21(4):306-318.
doi pubmed - Zhang Y, Wu KM, Yang L, Dong Q, Yu JT. Tauopathies: new perspectives and challenges. Mol Neurodegener. 2022;17(1):28.
doi pubmed - Alam J, Bhattacharjee S, Chakraborty S, Rahman SZ, Hasan A, Haseen MA, Sarfraz M. Neuromodulation via brain stimulation: A promising therapeutic perspective for Alzheimer’s disease. In: Ansari MA, Anand RS, Tripathi P, Mehrotra R, Heyat MBB (ed) Artificial intelligence in biomedical and modern healthcare informatics, 1st edn. Elsevier/Academic Press, London. 2024; p. 257-266.
- Misrani A, Tabassum S, Yang L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer's Disease. Front Aging Neurosci. 2021;13:617588.
doi pubmed - Blonz ER. Alzheimer's disease as the product of a progressive energy deficiency syndrome in the central nervous system: The Neuroenergetic Hypothesis. J Alzheimers Dis. 2017;60(4):1223-1229.
doi pubmed - Geldenhuys WJ, Van der Schyf CJ. Role of serotonin in Alzheimer's disease: a new therapeutic target? CNS Drugs. 2011;25(9):765-781.
doi pubmed - Claeysen S, Bockaert J, Giannoni P. Serotonin: a new hope in Alzheimer's disease? ACS Chem Neurosci. 2015;6(7):940-943.
doi pubmed - Angelucci ME, Cesario C, Hiroi RH, Rosalen PL, Da Cunha C. Effects of caffeine on learning and memory in rats tested in the Morris water maze. Braz J Med Biol Res. 2002;35(10):1201-1208.
doi pubmed - Wang H, Zhang H. Reconsideration of Anticholinesterase Therapeutic Strategies against Alzheimer's Disease. ACS Chem Neurosci. 2019;10(2):852-862.
doi pubmed - Wang R, Reddy PH. Role of Glutamate and NMDA Receptors in Alzheimer's Disease. J Alzheimers Dis. 2017;57(4):1041-1048.
doi pubmed - Brodsky M, Lesiak AJ, Croicu A, Cohenca N, Sullivan JM, Neumaier JF. 5-HT(6) receptor blockade regulates primary cilia morphology in striatal neurons. Brain Res. 2017;1660:10-19.
doi pubmed - Shi M, Chu F, Zhu F, Zhu J. Impact of anti-amyloid-beta monoclonal antibodies on the pathology and clinical profile of Alzheimer's disease: a focus on aducanumab and lecanemab. Front Aging Neurosci. 2022;14:870517.
doi pubmed - Alam J. Vitamins: a nutritional intervention to modulate the Alzheimer's disease progression. Nutr Neurosci. 2022;25(5):945-962.
doi pubmed - Alam J, Sharma L. Potential enzymatic targets in Alzheimer's: a comprehensive review. Curr Drug Targets. 2019;20(3):316-339.
doi pubmed - Gupta GL, Samant NP. Current druggable targets for therapeutic control of Alzheimer's disease. Contemp Clin Trials. 2021;109:106549.
doi pubmed - Ju Y, Tam KY. Pathological mechanisms and therapeutic strategies for Alzheimer's disease. Neural Regen Res. 2022;17(3):543-549.
doi pubmed - Cummings J, Lee G, Nahed P, Kambar M, Zhong K, Fonseca J, Taghva K. Alzheimer's disease drug development pipeline: 2022. Alzheimers Dement (N Y). 2022;8(1):e12295.
doi pubmed - Huang LK, Kuan YC, Lin HW, Hu CJ. Clinical trials of new drugs for Alzheimer disease: a 2020-2023 update. J Biomed Sci. 2023;30(1):83.
doi pubmed - Cummings J, Zhou Y, Lee G, Zhong K, Fonseca J, Cheng F. Alzheimer's disease drug development pipeline: 2023. Alzheimers Dement (N Y). 2023;9(2):e12385.
doi pubmed - Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci. 2020;27(1):18.
doi pubmed - Alam J, Jaiswal V, Sharma L. Screening of Antibiotics Against beta-amyloid as Anti-amyloidogenic Agents: A Drug Repurposing Approach. Curr Comput Aided Drug Des. 2021;17(5):647-654.
doi pubmed - Sharma L, Sharma A, Goyal R, Alam J. Pinus Roxburghii Sarg. Ameliorates Alzheimer’s disease - type neurodegeneration and cognitive deficits caused by intracerebroventricular-streptozotocin in rats: an in vitro and in vivo study. Indian JPharm Sci. 2020;82:861-870.
doi - Alvarez A, Munoz JP, Maccioni RB. A Cdk5-p35 stable complex is involved in the beta-amyloid-induced deregulation of Cdk5 activity in hippocampal neurons. Exp Cell Res. 2001;264(2):266-274.
doi pubmed - Ghosh A, Giese KP. Calcium/calmodulin-dependent kinase II and Alzheimer's disease. Mol Brain. 2015;8(1):78.
doi pubmed - Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C, Terro F. Tau protein kinases: involvement in Alzheimer's disease. Ageing Res Rev. 2013;12(1):289-309.
doi pubmed - Garcia-Alloza M, Tsang SW, Gil-Bea FJ, Francis PT, Lai MK, Marcos B, Chen CP, et al. Involvement of the GABAergic system in depressive symptoms of Alzheimer's disease. Neurobiol Aging. 2006;27(8):1110-1117.
doi pubmed - Louzada PR, Paula Lima AC, Mendonca-Silva DL, Noel F, De Mello FG, Ferreira ST. Taurine prevents the neurotoxicity of beta-amyloid and glutamate receptor agonists: activation of GABA receptors and possible implications for Alzheimer's disease and other neurological disorders. FASEB J. 2004;18(3):511-518.
doi pubmed - Calvo-Flores Guzman B, Vinnakota C, Govindpani K, Waldvogel HJ, Faull RLM, Kwakowsky A. The GABAergic system as a therapeutic target for Alzheimer's disease. J Neurochem. 2018;146(6):649-669.
doi pubmed - Rahman A. The role of adenosine in Alzheimer's disease. Curr Neuropharmacol. 2009;7(3):207-216.
doi pubmed - Albasanz JL, Perez S, Barrachina M, Ferrer I, Martin M. Up-regulation of adenosine receptors in the frontal cortex in Alzheimer's disease. Brain Pathol. 2008;18(2):211-219.
doi pubmed - Stockwell J, Jakova E, Cayabyab FS. Adenosine A1 and A2A receptors in the brain: current research and their role in neurodegeneration. Molecules. 2017;22(4):676.
doi pubmed - Santiago AR, Baptista FI, Santos PF, Cristovao G, Ambrosio AF, Cunha RA, Gomes CA. Role of microglia adenosine A(2A) receptors in retinal and brain neurodegenerative diseases. Mediators Inflamm. 2014;2014:465694.
doi pubmed - Albasanz JL, Rodriguez A, Ferrer I, Martin M. Adenosine A2A receptors are up-regulated in Pick's disease frontal cortex. Brain Pathol. 2006;16(4):249-255.
doi pubmed - Naddafi F, Mirshafiey A. The neglected role of histamine in Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2013;28(4):327-336.
doi pubmed - Colombres M, Sagal JP, Inestrosa NC. An overview of the current and novel drugs for Alzheimer's disease with particular reference to anti-cholinesterase compounds. Curr Pharm Des. 2004;10(25):3121-3130.
doi pubmed - Amat-Ur-Rasool H, Ahmed M, Hasnain S, Carter WG. Anti-cholinesterase combination drug therapy as a potential treatment for Alzheimer's disease. Brain Sci. 2021;11(2):184.
doi pubmed - Zhang Y, Li P, Feng J, Wu M. Dysfunction of NMDA receptors in Alzheimer's disease. Neurol Sci. 2016;37(7):1039-1047.
doi pubmed - Malinow R. New developments on the role of NMDA receptors in Alzheimer's disease. Curr Opin Neurobiol. 2012;22(3):559-563.
doi pubmed - Wulff BS, Hastrup S, Rimvall K. Characteristics of recombinantly expressed rat and human histamine H3 receptors. Eur J Pharmacol. 2002;453(1):33-41.
doi pubmed - Akincioglu H, Gulcin I. Potent acetylcholinesterase inhibitors: potential drugs for Alzheimer's disease. Mini Rev Med Chem. 2020;20(8):703-715.
doi pubmed - Hogan DB. Donepezil for severe Alzheimer's disease. Lancet. 2006;367(9516):1031-1032.
doi pubmed - Robinson DM, Keating GM. Memantine: a review of its use in Alzheimer's disease. Drugs. 2006;66(11):1515-1534.
doi pubmed - Liu J, Chang L, Song Y, Li H, Wu Y. The role of NMDA receptors in Alzheimer's disease. Front Neurosci. 2019;13:43.
doi pubmed - Wu C, Sun D. GABA receptors in brain development, function, and injury. Metab Brain Dis. 2015;30(2):367-379.
doi pubmed - Trinh PNH, Baltos JA, Hellyer SD, May LT, Gregory KJ. Adenosine receptor signalling in Alzheimer's disease. Purinergic Signal. 2022;18(3):359-381.
doi pubmed - Merighi S, Borea PA, Varani K, Vincenzi F, Travagli A, Nigro M, Pasquini S, et al. Pathophysiological role and medicinal chemistry of A(2A) adenosine receptor antagonists in Alzheimer's disease. Molecules. 2022;27(9):2680.
doi pubmed - Thangam EB, Jemima EA, Singh H, Baig MS, Khan M, Mathias CB, Church MK, et al. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: the hunt for new therapeutic targets. Front Immunol. 2018;9:1873.
doi pubmed - Lazewska D, Bajda M, Kaleta M, Zareba P, Doroz-Plonka A, Siwek A, Alachkar A, et al. Rational design of new multitarget histamine H(3) receptor ligands as potential candidates for treatment of Alzheimer's disease. Eur J Med Chem. 2020;207:112743.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.