| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://jnr.elmerpub.com |
Case Report
Volume 000, Number 000, July 2025, pages 000-000

From Brugada Syndrome to Sporadic Pseudopolyneuritic Amyotrophic Lateral Sclerosis
Pedro Enrique Labrada-Aguileraa, David Armando Guach-Heviab, e, Daniel Alejandro Gonzalez-Lagoc, Andres Luis Meneses-Roblesb, Laura Quinones-Martinezd, Armando Nielcen de la Rosa-Serranob
aDepartment of Neurology, “Lucia Iniguez Landin” Hospital, Holguin, Cuba
bDepartment of Neurology, Medical Sciences University, Holguin, Cuba
cDepartment of Neurosurgery, Medical Sciences University, Holguin, Cuba
dDepartment of Anesthesiology, Medical Sciences University, Holguin, Cuba
eCorresponding Author: David Armando Guach-Hevia, Department of Neurology, Medical Sciences University, Holguin, Cuba
Manuscript submitted January 16, 2025, accepted June 18, 2025, published online July 8, 2025
Short title: Pseudopolyneuritic Sporadic ALS
doi: https://doi.org/10.14740/jnr1015
| Abstract | ▴Top |
Amyotrophic lateral sclerosis is a fatal neurodegenerative disease characterized by progressive death of the upper and lower motor neurons in the central nervous system. Death in these patients has been associated with respiratory muscle weakness, while cardiovascular causes of mortality have been rarely reported. Brugada syndrome is a potentially lethal dysrhythmia that has been seldomly diagnosed in these patients, with only one case reported so far. We present a case of a patient with type 1 Brugada syndrome that was diagnosed with sporadic pseudopolyneuritic amyotrophic lateral sclerosis subtype. A 38-year-old black male patient, with a previous diagnosis of type 1 Brugada syndrome, came into our service complaining of progressive distal strength decreasing in his left leg, involuntary contractions of the muscles of his arms and legs, and body weight loss. The electrocardiogram described an elevation of J point and ST-segment of approximately 3 mm in all right precordial leads (V1 to V3), with negative T-wave in V1 lead, and incomplete right bundle branch blockade pattern. On neurological examination, left leg muscular weakness, global hyporeflexia, and atrophy of different muscles were found. Needle electromyogram detected fibrillations at rest in the left tibialis anterior muscle and abundant fibrillations and sharp waves in the left internal gastrocnemius and left femoral vastus muscles. A sporadic pseudopolyneuritic ALS subtype diagnosis was made. Multidisciplinary treatment was indicated. A stress-free life, change of labor, healthy diet, and daily physiotherapy sessions were oriented. Symptomatic treatment was prescribed with baclofen, carbamazepine, truabin, and folic acid. The Brugada syndrome was treated with an automatic implantable cardioverter-defibrillator. At 3 months evaluation, the clinical improvement was minimum. A full-time caregiver was recommended. ALS is a multisystem neurodegenerative disorder, with disease heterogeneity at clinical, genetic, and neuropathological levels. Cardiovascular causes, especially malignant arrhythmias such as Brugada syndrome, can also be associated with these patients. Further studies directed to analyze the potential relation of these two entities should be performed. Periodic surveillance with electrocardiogram to identify patterns of this dysrhythmia in ALS patients can be hereafter considered.
Keywords: Amyotrophic lateral sclerosis; Brugada syndrome; Cuba
| Introduction | ▴Top |
Amyotrophic lateral sclerosis (ALS) is defined by escalating decline of the upper motor neuron (UMN) and lower motor neuron (LMN) in the brain and spinal cord, conducing to muscle failing, paralysis and, finally, death due to respiratory failure within 3 - 5 years after the onset of symptoms [1]. Its incidence has been described to be between 0.6 and 3.8 per 100,000 person-years [2]. About 90% of cases are sporadic amyotrophic lateral sclerosis (sALS), and the extra 10% cases are familial [3]. Risk factors of sALS include advanced age (between 55 and 65 years) [4], male gender, genetic background, smoking, head injury, body mass index, viral infections, somatic stress, and exposure to heavy metals, constant pollutants, and environmental toxins such as methylamino-L-alanine [5]. The diagnosis of ALS remains mostly clinical and is suggested on the existence of both UMN and LMN signs, in patients with escalating muscle weakness in whom no different justification can be found [6]. The initial symptoms due to motor neuronal degeneration are fasciculation, muscle cramps and stiffness, dysarthria, sialorrhea, dysphagia, and emotional instability characterized by pseudobulbar affect and dyspnea [7]. In addition, electrodiagnostic tests with needle electromyogram (EMG) and neuroimaging are truly helpful to corroborate the diagnosis [8]. Although great pharmacological advances have been made with riluzole and edaravone by using molecular hybridization [1], there is still no cure available. Therefore, current treatment remains mostly symptomatic, and its success depends on a multidisciplinary approach that includes pharmacological and non-pharmacological interventions [6].
Death in these patients has been associated with respiratory muscle weakness, while cardiovascular causes of mortality have been rarely reported. Cardiac failure due to Brugada syndrome (BS) has been eventually related with different types of motor neuron diseases [9], though its specific diagnosis in ALS patients is very rare, with only one case report available in the literature [10].
BS is a rare inherited disorder associated with the risk of ventricular fibrillation and sudden cardiac death (SCD) with a characteristic electrocardiogram (ECG) (coved type ST-segment elevation 2 mm followed by a negative T-wave in one of the right precordial leads V1 to V3) [11].
This condition is responsible for > 10% of all SCDs, with a prevalence 8 - 10 times higher in men than in women, and syncope is the typical clinical presentation, though most patients remain asymptomatic [12].
We present a case of a 38-year-old black male patient with a clinical background of sporadic sudden loss of consciousness that had been followed since 2018 by the cardiology department. He was finally diagnosed with type 1 BS 6 months before our study, and just 2 months later developed progressive lower limbs weakness and involuntary contractions, along with other symptoms of motor compromise. After examination and abnormal EMG, a diagnosis of sALS was made, and proper multidisciplinary treatment was indicated for both conditions. To date, these two entities have never been described in Cuba in a same patient, and there are very few reports about ALS with cardiac involvement worldwide.
| Case Report | ▴Top |
Investigations
A 38-year-old non-married black male patient, right-handed, gardener, came into our service complaining of progressive distal weakness in his left leg and involuntary contractions of the muscles of his arms and legs, associated with significant body weight loss of approximately 17 kg that started about 4 months before presentation. On past medical history, he had possible BS followed by cardiology, with episodes of sudden loss of consciousness since 2018 that were accompanied by sweating, paleness, chest discomfort, and arterial hypotension related to periods of prolonged fasting, intense physical activity, or emotional stress. The last one of these events occurred just 6 months before as reported in his personal clinical file, with a diagnostic type 1 BS’s ECG describing an elevation of both J point and ST-segment of approximately 3 mm in all right precordial leads (V1 to V3), with negative T-wave in V1 lead, and a pattern of incomplete right bundle branch blockade (Fig. 1). After this, an automatic implantable cardioverter-defibrillator (ICD) placement was recommended, but he declined the recommendation and signed the proper patient’s consent. At the moment of this consultation, the patient remained untreated by his own decision. He denied smoking or drinking. On family history, his mother suffered from high blood pressure and had a sicklemic trait.
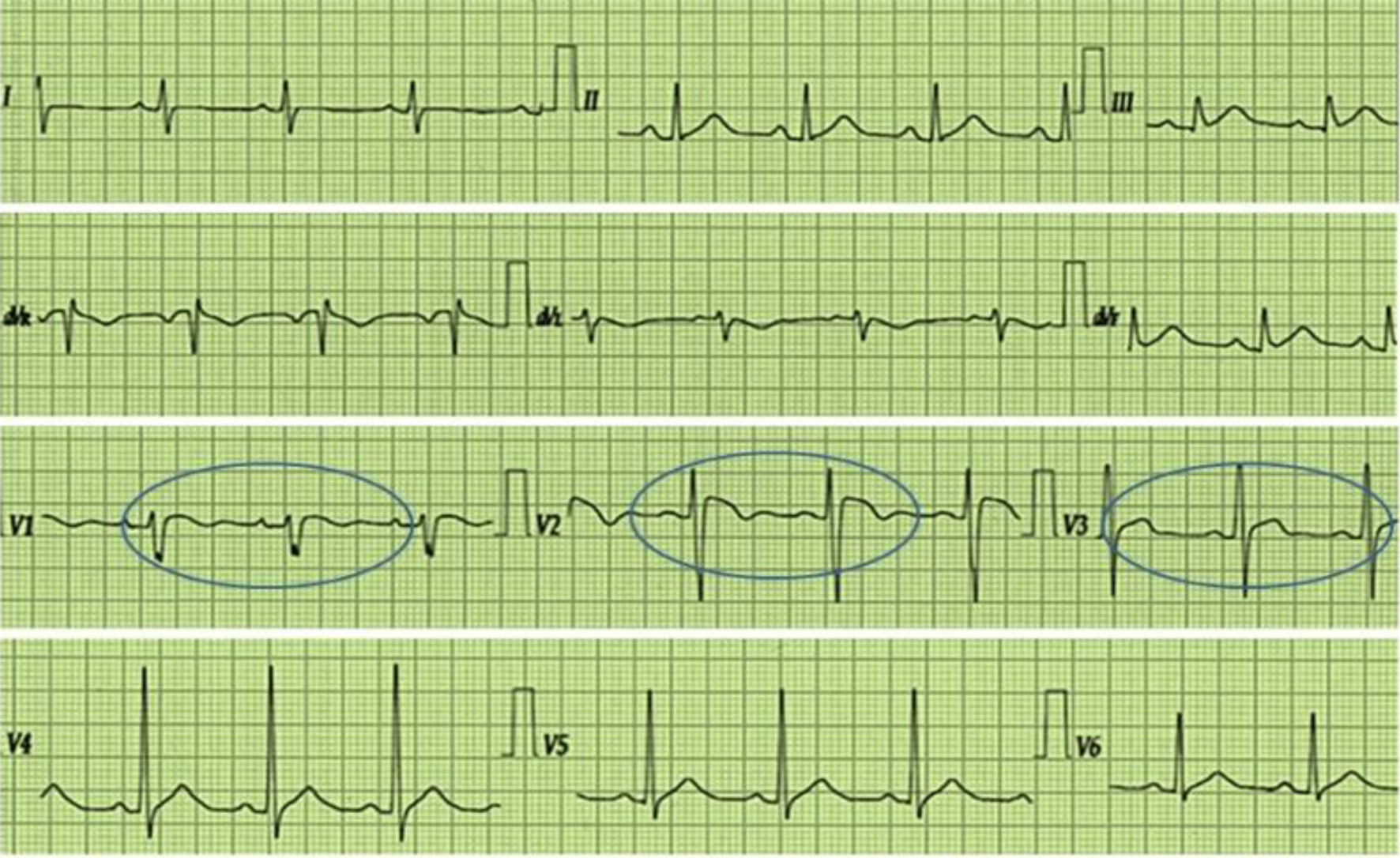 Click for large image | Figure 1. The image shows a standard 12-lead electrocardiogram describing an elevation of both J point and ST-segment of approximately 3 mm in all right precordial leads (V1 to V3), with negative T-wave in V1 lead, and a pattern of incomplete right branch blockade. The circles highlight the alterations. |
On physical exam, no cardiovascular alterations were found and physiological parameters were normal. Neurological examination applying the modified Medical Research Council muscular strength scale showed a score of 3/5 with movement only against gravity in his left leg. Axial muscles, the right leg, and upper limbs evaluation showed a 5/5 score. The National Institute of Neurological Disorders and Stroke (NINDS grading) scale showed a +1 score due to global hyporeflexia. Sensitivity was preserved in all sensory modalities. Bilateral Hoffman, Tromner, and Babinski signs were present.
In addition, atrophy of the masseter, temporal, left quadriceps femoris, and gastrocnemius muscles was described along with thenar, hypothenar, and interosseous atrophy in both hands. Furthermore, spontaneous lingual fasciculations, nauseous areflexia, and high arched feet were observed.
Blood studies showed no alteration. Unfortunately, genetic and biomarker’s surveillance was not available at the moment of consultation. In addition, a Holter study was indicated, describing a ventricular repolarization disorder of autonomic etiology with ST-segment elevation of 3 mm with superior concavity in channel 1, simultaneous ST in the isoline in channel 2, and sudden changes in the heart rate even with normal sinus rhythm. Echocardiography showed no current alteration.
Brain and spinal cord magnetic resonance imaging were normal. On the other hand, electroneurogram showed motor nerve fiber axonal damage and de-myelination of left median nerve, with additional axonal damage of nerve fibers of the bilateral deep peroneal nerve. Needle EMG detected spontaneous isolated fibrillations at rest in the left tibialis anterior muscle and abundant fibrillations and sharp waves in the left internal gastrocnemius muscle and left femoral vastus muscle, with large motor unit potentials of increased duration (Fig. 2).
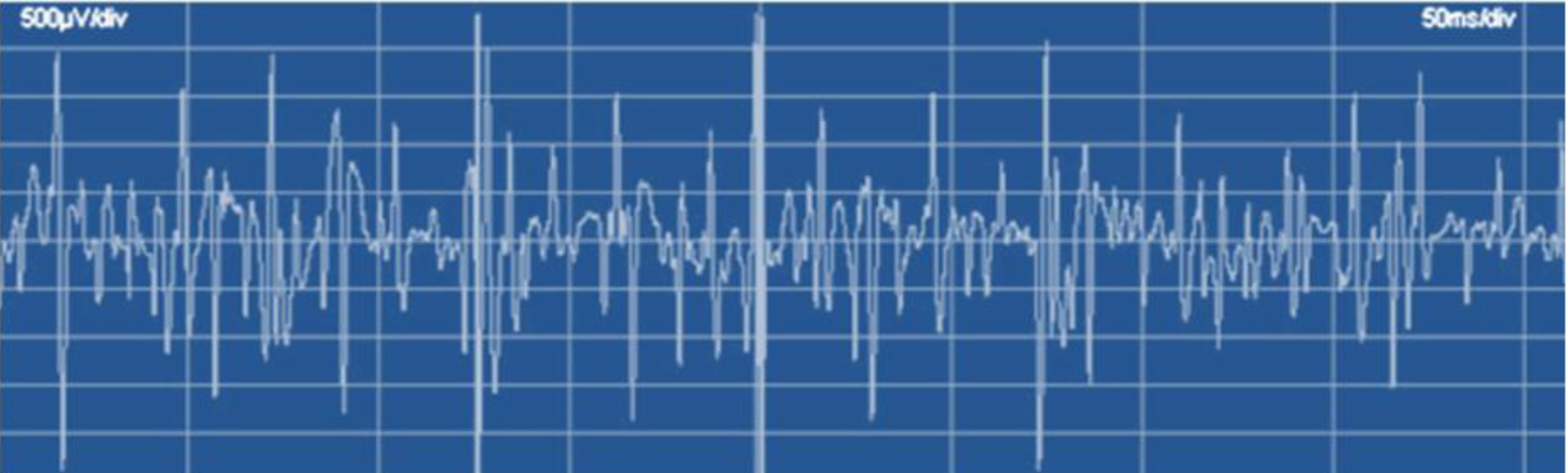 Click for large image | Figure 2. Needle electromyogram of the left internal gastrocnemius muscle. Note abundant fibrillations and sharp waves, with poor full contraction isolated pattern. |
Furthermore, EMG of the tongue showed spontaneous activity at rest represented by fasciculations and fibrillations in both sides of the tongue. Well-established large motor unit potentials of increased amplitude with polyphasic motor unit recruitment were concluded.
Diagnosis
Based on the clinical history and EMG data, an sALS diagnosis was made, and it was classified as a pseudopolyneuritic subtype. As differential diagnoses, chronic inflammatory demyelinating polyneuropathy, Steinert myotonic dystrophy, primary progressive multiple sclerosis, hereditary Charcot-Marie-Tooth type 1 polyneuropathy, oculopharyngeal dystrophy, and posterior territory cerebrovascular disease were considered. All subtypes of ALS were also ruled out.
Treatment
After the final diagnosis, in the presence of the cardiologist and the patient’s family members, the patient was introduced to the particularities of his newly diagnosed disease, and a multidisciplinary follow-up by the cardiology and neurology services, along with psychologists, dietary advisors, and physiotherapists was started.
A stress-free life, a change of labor, and a quiet family and recreational environment were the first suggestions. A healthy diet rich in fruits and vegetables, with a frequent consumption of prebiotic and probiotic based food was also oriented. In addition, twice daily home sessions of stretching, weight bearing, and muscle massaging performed by his relatives were prescribed. Symptomatic pharmacologic treatment was also indicated, with baclofen (10 mg) 0.5 tablet each 8 h, carbamazepine (200 mg) 0.5 tablet each 12 h [13], Truabin 1 weekly intramuscular ampoule for 10 weeks, and folic acid (5 mg) one tablet daily. Furthermore, the patient was once again warned about the risks of having his BS untreated, and possibly under the stimuli of the new diagnosis, he finally agreed on receiving an automatic ICD that was placed 4 days later by the cardiologist.
Follow-up and outcomes
At 3 months evaluation, the patient’s clinical improvement was minimum, though he achieved some independence for daily living activities. No new syncope event was reported. A full-time caregiver was recommended.
| Discussion | ▴Top |
ALS is a destructive neurodegenerative illness in adults that was firstly recorded by the neurobiologist Jean-Martin Charcot in 1869. This condition is also called Lou Gehrig’s disease in the United States, to pay tribute to the baseball player who was diagnosed with it in the 1930s [14]. Formerly described as a pure motor neuron illness by Charcot, it is currently known as a multisystem neurodegenerative disorder, with disease heterogeneity at the clinical, genetic, and neuropathological levels [6]. Our patient age at onset was relatively early, considering that average age of onset is after the fifth decade of life [4].
Risk factors identified on the patient were male gender, physical stress, and a possible exposure to environmental toxins considering that he had been a gardener for almost a decade. Neither genetic testing nor biomarker survey was available at the moment of consultation, which was an objective limitation. The five most prevalent genes tested in ALS are C9orf72, SOD1, TDP-43, FUS, and TBK-1 [6]. However, no family positive history for ALS was referred, hence sALS diagnosis was made. Based on the current literature, there is no direct evidence establishing a causal relationship between ALS and BS. However, emerging genetic and molecular research suggests potential mechanistic overlaps, particularly involving shared genes and pathways. In a study of BS patients, Jeong et al identified pathogenic variants in DSP, a gene encoding a desmosomal protein critical for cell-cell adhesion [15]. DSP mutations are classically linked to arrhythmogenic cardiomyopathy through sodium channel disruption, but have also been implicated in ALS pathogenesis through disrupted cytoskeletal integrity in motor neurons. Other genes like PKP2 (plakophilin-2), SCN5A, SCN10A 12, CACNA1C (L-type calcium channel), TRPM4, GPD1L, SOD1, and C9orf72 also provide a rationale for further study of overlapping between these two entities [15, 16], considering that all these genes influence either the beginning or progression of both diseases.
Based on clinical and needle EMG findings, our patient was diagnosed with a pseudopolyneuritic form of ALS, also called distal flail leg syndrome or leg amyotropic diplegia, firstly described in the thesis of Jean Patrikios in 1918, supervised by Pierre Marie [17]. According to Grad et al, it is a relatively rare subtype of ALS, representing 3-3.5% of all motor neuron disease cases, and occurs mostly in men and largely in the LMN, with slow clinical evolution, mean survival ranging from 76 to 96 months [3], weak or absent lower limb deep tendon reflexes, and progressive asymmetrical leg distal weakness [17].
Regarding diagnosis, it is fundamental to highlight the BS’s previous follow-up of our patient. BS is an inherited arrhythmia syndrome described by coved (type 1) or saddleback (types 2 and 3) ST-segment elevations (≥ 2 mm), continued by deep T-wave inversions in leads V1-V3, related to augmented threat of SCD [10]. This syndrome can induce ventricular tachyarrhythmia and circulatory collapse, and to the best of our knowledge, it has been noticed only once in a patient with ALS. Battineni et al in their case report described a middle-aged female patient with recent sALS that was diagnosed with type 2 BS and eventually suffered from pulseless ventricular tachycardia and fibrillation, requiring intense resuscitation management [10].
Although the association between ALS and BS is very rare and yet not fully understood, several studies have described specific cardiac involvement in ALS morbidity and mortality. In a French ALS study by Gil et al, who researched the main causes of mortality, 3.4% of patients died due to myocardial infarction or dysrhythmias, and a few patients had SCD [18]. Cardiovascular aberrations such as QT prolongation, heart rate blood pressure dyssynchrony, and nocturnal hypotension have been frequently reported in ALS patients [10]. The progressive decrease of the sympathetic tone secondary to degeneration of the intermediolateral column in the upper levels of the spinal cord could be the cause behind this event, as previously shown in pathological studies by Asai et al [19]. Additionally, a systematic review made by Xu et al showed that cardiovascular comorbidities in ALS patients vary importantly when comparing different regions, finding that the comorbidity of hypertension in France (56.9%) was the highest, followed by Portugal (48%), America (25%), and China (15%), and only 4-5% of patients with ALS in Australia or the Netherlands experienced coronary heart disease [20]. The major results of their study concluded that hypertension could reduce the survival of ALS, coronary heart disease could increase the risk of ALS, and heart failure represents a negative prognostic factor for ALS [20].
Currently, there is a hypothesis that patients with ALS are at high risk for BS pattern, regarding the efferent sympathetic dysfunction shared with Kennedy’s disease [10]. Further studies directed to analyze the potential relation of these two entities should be performed. Continued surveillance with ECG to identify patterns of BS in ALS patients should be considered in order to diagnose this dangerous and potentially lethal dysrhythmia in the course of such a complex neurodegenerative disorder.
Finally, treatment was indicated based on a multidisciplinary approach. Both pharmacological and non-pharmacological interventions are valid. Spasticity can be treated with baclofen, tizanidine, cannabinoids, and muscle stretching, and sialorrhea can be treated with anticholinergic medications (amitriptyline, glycopyrronium bromide, and oxybutynin) and botulin toxin injections into the salivatory glands. Muscle cramps may respond to magnesium supplements, quinine sulfate, gabapentin, or carbamazepine. A selective serotonin reuptake inhibitor, amitriptyline, benzodiazepines and dextromethorphan hydrobromide/quinidine sulfate, can be used if emotional lability [6]. Additionally, dietary changes can improve nutrition. For instance, according to Almaguer-Mederos et al, prebiotic and probiotic based food is highly recommended for patients suffering from different types of neurodegeneration, including ALS, due to its influence on the gut microbiota brain axis [21]. Furthermore, a gastrostomy tube can be considered if the caloric intake is deficient or when swallowing becomes difficult. Speech therapy is commonly required and assisted communication (customized software) can also help. Non-invasive ventilation is the main life-prolonging treatment for respiratory insufficiency. Patient’s individual wishes should be taken into account along the process and advance care planning should be initiated early [6].
New therapeutic approaches headed by molecular hybridization, in which different agents can be used simultaneously on several targets, can give a new hope in the treatment of ALS. Specifically, riluzole and edaravone, approved drugs for the treatment of ALS, attract great debate for the molecular hybridization approach [1]. None of these pharmacological therapies are available in our country.
Considering BS, lifestyle changes with education, ICD implantation, quinidine, and catheter ablation of epicardial late activation areas in the right ventricle are proven accurate approaches for the prevention of SCD. In addition, all BS patients should be alerted of the conditions, medications, and substances that might induce ventricular arrhythmias [22].
Conclusions
ALS is a multisystem neurodegenerative disorder, with disease heterogeneity at the clinical, genetic, and neuropathological levels. Cardiovascular causes, especially malignant arrhythmias such as BS, can also be linked to patients with ALS. Further studies directed to analyze the potential relation of these two entities should be performed. Continued surveillance with ECG to detect patterns of BS in ALS patients can be considered in patients with cardiac symptoms in order to diagnose this dangerous and potentially lethal dysrhythmia in the course of such a complex neurodegenerative disorder.
Acknowledgments
We would like to express our sincere gratitude to the patient and his family for their support and trust throughout this process.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Written informed consent was obtained from the patient to publish this case report.
Author Contributions
Conception, design and literature searches: DAGH, ALMR, LQM, ANRS; Data collecting: PELA, DAGL; Drafting the article: DAGH; Critical revision of the manuscript for important intellectual content: PELA, DAGL.
Data Availability
The data that support the findings of this study are available from the corresponding author on request.
| References | ▴Top |
- Sever B, Ciftci H, DeMirci H, Sever H, Ocak F, Yulug B, Tateishi H, et al. Comprehensive research on past and future therapeutic strategies devoted to treatment of amyotrophic lateral sclerosis. Int J Mol Sci. 2022;23(5):2400.
doi pubmed - Longinetti E, Fang F. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol. 2019;32(5):771-776.
doi pubmed - Grad LI, Rouleau GA, Ravits J, Cashman NR. Clinical spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb Perspect Med. 2017;7(8):a024117.
doi pubmed - Byrne S, Jordan I, Elamin M, Hardiman O. Age at onset of amyotrophic lateral sclerosis is proportional to life expectancy. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(7-8):604-607.
doi pubmed - Farace C, Fenu G, Lintas S, Oggiano R, Pisano A, Sabalic A, Solinas G, et al. Amyotrophic lateral sclerosis and lead: A systematic update. Neurotoxicology. 2020;81:80-88.
doi pubmed - Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27(10):1918-1929.
doi pubmed - Martinez A, Palomo Ruiz MD, Perez DI, Gil C. Drugs in clinical development for the treatment of amyotrophic lateral sclerosis. Expert Opin Investig Drugs. 2017;26(4):403-414.
doi pubmed - Shefner JM, Al-Chalabi A, Baker MR, Cui LY, de Carvalho M, Eisen A, Grosskreutz J, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. 2020;131(8):1975-1978.
doi pubmed - Finsterer J, Stollberger C, Maeztu C. Sudden cardiac death in neuromuscular disorders. Int J Cardiol. 2016;203:508-515.
doi pubmed - Battineni A, Gummi R, Mullaguri N, Govindarajan R. Brugada syndrome in a patient with amyotrophic lateral sclerosis: a case report. J Med Case Rep. 2017;11(1):192.
doi pubmed - Brugada J, Campuzano O, Arbelo E, Sarquella-Brugada G, Brugada R. Present status of Brugada syndrome: JACC State-of-the-Art review. J Am Coll Cardiol. 2018;72(9):1046-1059.
doi pubmed - Pappone C, Santinelli V. Brugada syndrome: progress in diagnosis and management. Arrhythm Electrophysiol Rev. 2019;8(1):13-18.
doi pubmed - Xu X, Shen D, Gao Y, et al. A perspective on therapies for myotrophic lateral sclerosis: can disease progression be curbed? Transl Neurodegener. 2021;10:29.
doi - Yang X, Ji Y, Wang W, Zhang L, Chen Z, Yu M, Shen Y, et al. Amyotrophic lateral sclerosis: molecular mechanisms, biomarkers, and therapeutic strategies. Antioxidants (Basel). 2021;10(7):1012.
doi pubmed - Jeong JH, Lee HS, Choi YY, et al. Clinical role of genetic testing for the Brugada syndrome overlapping with arrhythmogenic cardiomyopathy. Int J Arrhythm. 2024;25:12.
doi - Pan Jie, Hu KX, Yao WL. Some advances on genetics related to Brugada syndrome. Austin Cardio & Cardiovasc Case Rep. 2016;1(1):1004.
- Wijesekera LC, Mathers S, Talman P, Galtrey C, Parkinson MH, Ganesalingam J, Willey E, et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology. 2009;72(12):1087-1094.
doi pubmed - Gil J, Funalot B, Verschueren A, Danel-Brunaud V, Camu W, Vandenberghe N, Desnuelle C, et al. Causes of death amongst French patients with amyotrophic lateral sclerosis: a prospective study. Eur J Neurol. 2008;15(11):1245-1251.
doi pubmed - Asai H, Hirano M, Udaka F, Shimada K, Oda M, Kubori T, Nishinaka K, et al. Sympathetic disturbances increase risk of sudden cardiac arrest in sporadic ALS. J Neurol Sci. 2007;254(1-2):78-83.
doi pubmed - Xu K, Ji H, Hu N. Cardiovascular comorbidities in amyotrophic lateral sclerosis: A systematic review. J Clin Neurosci. 2022;96:43-49.
doi pubmed - Almaguer Mederos LE, Martinez Martinez W, Guach Hevia D. Implicaciones de la microbiota intestinal en la etiologia y terapeutica de la enfermedad de Parkinson. Rev Haban Cienc Med. 2018;17(1):48-57.
- Bayrak F, Brugada P. Recent status in Brugada syndrome. Turk Kardiyol Dern Ars. 2022;50(2):137-144.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.