| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://jnr.elmerpub.com |
Case Report
Volume 000, Number 000, March 2025, pages 000-000

Late-Onset CLCN2-Related Leukodystrophy Associated With A Novel Mutation
Slaven Pikijaa, Julia Feigea, Andreea Tomaa, Richard Radlbergera, Eugen Trinkaa, b, c, Fritz Klausnerd, Mahdi Safdariana, e
aDepartment of Neurology, Neurocritical Care and Neurorehabilitation, Christian Doppler University Hospital, Centre of Cognitive Neuroscience, Paracelsus Medical University, Member of EpiCARE, Salzburg, Austria
bNeuroscience Institute, Christian Doppler University Hospital, Centre of Cognitive Neuroscience, Paracelsus Medical University, Salzburg, Austria
cKarl Landsteiner Institute for Neurorehabilitation and Space Neurology, Salzburg, Austria
dDepartment of Neuroradiology, Christian Doppler University Ho
spital, Paracelsus Medical University, Salzburg, Austria
eCorresponding Author: Mahdi Safdarian, Department of Neurology, Neurocritical Care and Neurorehabilitation, Christian Doppler University Hospital, Centre of Cognitive Neuroscience, Paracelsus Medical University, Member of EpiCARE, Salzburg, Austria
Manuscript submitted December 16, 2024, accepted February 26, 2025, published online March 10, 2025
Short title: Late-Onset CLCN2 Leukodystrophy With Novel Mutation
doi: https://doi.org/10.14740/jnr866
| Abstract | ▴Top |
Loss-of-function mutations in the chloride channel protein 2 (CLCN2) gene cause defective channel function, leading to a specific type of leukodystrophy named CLCN2-related leukoencephalopathy. This is a rare disease that can become apparent either in childhood or adulthood but generally does not progress significantly. Here, we present an infertile man in his 60s with genetically confirmed novel homozygotic CLCN2 mutation and typical magnetic resonance imaging (MRI) findings of leukodystrophy who presented with severe headache to our clinic. The patient had no other specific neurologic findings, and we believe he is the oldest case reported in the literature with genetically confirmed diagnosis of CLCN2-related leukodystrophy.
Keywords: Chloride channel protein 2; Leukoencephalopathy; Leukodystrophy
| Introduction | ▴Top |
Chloride channel 2 (ClC-2) is a voltage-gated channel with a broad expression profile, playing a crucial role in various physiological processes, including ion homeostasis and cell volume regulation [1]. ClC-2, like other CLC family members, is involved in the control of electrical excitability, intracellular ion balance, and transepithelial transport, contributing to essential cellular and physiological functions [2]. Mutations in the chloride channel protein 2 (CLCN2) gene can lead to distinct clinical conditions depending on the nature of the mutation. Loss-of-function mutations are associated with CLCN2-related leukoencephalopathy, a specific type of leukodystrophy [3]. In contrast, gain-of-function mutations in CLCN2 have been linked to hyperaldosteronism, a condition characterized by excessive aldosterone production and hypertension [4, 5]. This is a rare disease and can become apparent either in childhood or in adults but generally does not progressively worsen. Those diagnosed as children may have learning difficulties, while vision problems are more common in those with symptoms beginning in adulthood [6]. Some nonspecific symptoms such as ataxia and frequent headaches have been also reported, in addition to male infertility. Affected individuals have characteristic findings in brain magnetic resonance imaging (MRI) including intramyelinic edema [7]. Leukodystrophies are a genetically determined group of diseases characterized by progressive degeneration of the white matter, caused by impaired development of the myelin sheath leading to various clinical phenotypes [6].
Here, we present an infertile man in its 60s with genetically confirmed novel homozygotic CLCN2 mutation and typical MRI findings of leukodystrophy who presented with severe headache to our clinic. The patient had no other specific neurologic findings, and we believe he is the oldest case reported in the literature with genetically confirmed diagnosis of CLCN2-related leukodystrophy.
| Case Report | ▴Top |
A 64-year-old man presented to our neurological emergency due to a severe sudden stabbing headache, visual analog scale 7 of 10 (10 depicting subjectively worst imaginable headache), nausea and repeated vomiting, and difficulty in finding words, lasting for about 20 min. He presented at admission with headache and recurrent vomiting without focal neurological deficits. He was under treatment with citalopram for mild depression. The patient was born from a consanguineous marriage (grandmothers of the patient were sisters) and had one sister who is clinically without symptoms. A brain MRI, using fluid-attenuated inversion recovery (FLAIR) sequences, revealed symmetrical hyperintensities in both middle cerebellar peduncles, the pons, and a large part of the posterior limbs of the internal capsules. In the area of the precentral region, only discrete hyperintense changes in the FLAIR sequence were detected on both sides (Fig. 1).
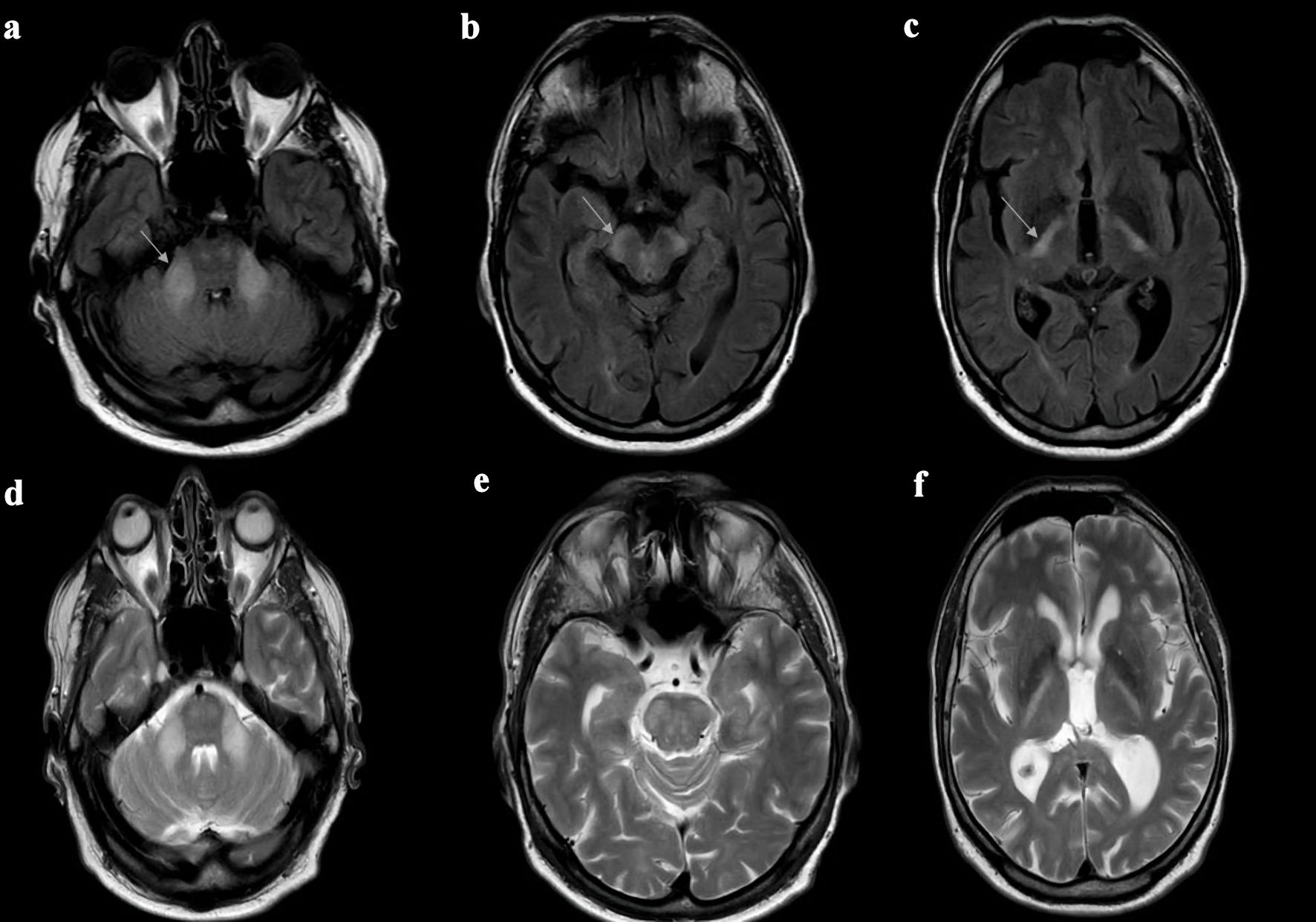 Click for large image | Figure 1. Axial brain MRI FLAIR sequences (a-c) and T2-weighted sequences (d-f) of the 64-year-old patient. Both axial MRI FLAIR and T2-weighted sequences of the patient demonstrate abnormal hyperintensities in the middle cerebellar peduncles (a, d), cerebral peduncles (b, e) and posterior limb of internal capsule (c, f). This type of leukodystrophy, affecting the white matter of the brain (including the internal capsule and cerebellar peduncles), is observed in patients with CLCN2-related leukoencephalopathy (CC2L). Thus, gene analysis was conducted and confirmed the diagnosis. MRI: magnetic resonance imaging; FLAIR: fluid-attenuated inversion recovery. |
Electroencephalography (EEG) results were normal. Cardiac evaluation, including electrocardiogram (EKG) and echocardiography, was normal. Goldmann perimetry showed a concentrically narrowed field of view. Optical coherent tomography showed no abnormal findings. On the urological evaluation, azoospermia was described. Initially, hypokalemia was documented, which, after oral substitution returned to normal at the time of discharge. In particular, with regard to paraneoplastic or neuronal antibodies, the findings were unremarkable. Based on FLAIR imaging, genetic evaluation was initiated and revealed homozygous novel pathogenic CLCN2 mutation (exon 3: c.318G>A, p.W106*-Class 5), confirming clinical suspicion of leukodystrophy.
| Discussion | ▴Top |
Gene description
The CLCN2 gene, located on chromosome 3q27 (Online Mendelian Inheritance in Man (OMIM) = 600570.0006), belongs to the CLC gene family, which encodes chloride channels essential for maintaining cellular chloride homeostasis. This channel’s activity is considered particularly critical in neurons within the brain [2]. Sik et al demonstrated that CLCN2 channels are localized in the plasma membranes of dendrites, axons, and somata of both pyramidal and nonpyramidal cells in the hippocampus [8]. However, while CLCN2 may be expressed in neurons, its confirmed physiological role appears to be in glial cells. Depienne et al [6], using immunohistochemistry on healthy human brain tissue, observed CLCN2 expression on the surface of cell bodies and processes of nearly all glial fibrillary acidic protein (GFAP)-positive fibrous astrocytes in the posterior limb of the internal capsule. They also identified CLCN2 expression along axons, in oligodendrocytes, and within the ependymal lining [6]. Notably, CLCN2 function in glial cells is modulated by GlialCAM, a glial adhesion molecule that acts as an essential regulatory subunit of ClC-2, altering its localization and biophysical properties [9]. Furthermore, studies in mouse models have demonstrated that Clcn2 mutations can lead to leukodystrophy and vacuolation in myelin-rich regions, emphasizing the channel’s critical role in glial function and central nervous system (CNS) homeostasis [10].
Electron microscopy further confirmed CLCN2 localization in white matter astrocytes, particularly in astrocyte processes involved in astrocyte-astrocyte and astrocyte-abaxonal myelin contacts [6]. Mutations in the CLCN2 gene result in distinct pathologies based on their functional impact. Loss-of-function mutations disrupt chloride ion transport, leading to CLCN2-related leukoencephalopathy, characterized by white matter abnormalities and neurological symptoms [6]. Conversely, gain-of-function mutations enhance chloride conductance, causing hyperaldosteronism. This condition is marked by increased aldosterone synthesis, leading to hypertension and electrolyte imbalances. The identification of these mutations underscores the pivotal role of ClC-2 channels in both neural integrity and endocrine regulation [4]. Mutations in the CLCN2 gene cause certain brain cells and the myelin (white matter) surrounding neurons to accumulate water, impairing nerve impulse transmission and resulting in neurological symptoms such as ataxia and other features of CLCN2-related leukoencephalopathy. To date, at least 18 mutations in the CLCN2 gene have been identified as causative for CLCN2-related leukoencephalopathy [11].
The CLCN2 variant c.318G>A, p.W106*, has not been previously described as pathogenic, and is present at a very low allele frequency in the gnomAD control database. The variant is a stop variant, which causes the mutant mRNA either to degrade prematurely, leading to a loss of function of the affected allele or to a truncated protein with reduced or aberrant function. Homozygous or compound-heterozygous pathogenic variants in the CLCN2 gene were associated with leukodystrophy with ataxia (OMIM 615651), and there have already been numerous patients with homozygous or compound-heterozygous loss-of-function mutations described [7]. We therefore evaluate the CLCN2 variant exon 3: c.318G>A, p.W106*-Class 5, as pathogenic.
Clinical and neuroradiological findings
Leukoencephalopathy with ataxia (LKPAT; 615651) is a distinct neurological disorder characterized by a specific pattern of white matter abnormalities on brain MRI. Research indicates that this condition is caused by homozygous or compound heterozygous mutations in the CLCN2 gene located on chromosome 3q27. Brain MRI findings include prominent signal abnormalities and reduced apparent diffusion coefficient (ADC) values in regions such as the posterior limbs of the internal capsules, middle cerebral peduncles, pyramidal tracts in the pons, and middle cerebellar peduncles, along with evidence of myelin microvacuolation [6].
Neurologic findings consist of a wide variety of nonspecific symptoms including gait instability (ataxia) following initially normal motor development, as well as frequent headaches and mild spasticity. Cognitive deficits, psychiatric manifestations (such as depression and schizophrenia-like symptoms), and learning challenges may also be present. Some patients experience auditory issues, including hearing loss, tinnitus, and vertigo. Male patients with this condition may have infertility, as observed in our case. Affected individuals typically retain the ability to walk unaided, do not require mobility support, and rarely experience blindness [12]. The diagnosis of CLCN2-related leukoencephalopathy is confirmed through molecular genetic testing that identifies bi-allelic pathogenic variants in CLCN2 [12].
CLCN2-related leukoencephalopathy follows an autosomal recessive inheritance pattern. Assessing the genetic status of both older and younger siblings of the proband is advisable to enable early identification of those, who might benefit from prompt diagnosis and regular monitoring for motor, cognitive, visual, and auditory impairments [12]. Routine neurologic evaluations annually, as well as ophthalmologic and audiologic assessments every 2 - 3 years, are also recommended.
Previous studies
Azoospermia, or the absence of sperm in semen, has been linked to loss-of-function mutations in the CLCN2 gene. Studies using Clcn2 knockout (KO) mouse models have provided insights into the underlying mechanisms. Bosl et al discovered that deleting the Clcn2 gene in mice causes severe degeneration in the retina and testes, resulting in male-specific infertility [13]. They demonstrated that male Clcn2-/- mice exhibit infertility due to a significant loss of germ cells, leading to azoospermia [13]. Further research by Blanz et al revealed that the absence of ClC-2 channels in Sertoli cells, which support and nourish developing sperm, disrupts the microenvironment essential for spermatogenesis, thereby resulting in male infertility [14]. These findings underscore the critical role of ClC-2 channels in maintaining male fertility. They also reported that these mice exhibited blindness and gradually developed extensive spongiform vacuolation in the white matter of both the brain and spinal cord [14]. Fluid-filled cavities formed between the myelin sheaths in the CNS, whereas the peripheral nervous system remained unaffected. Despite this, neuronal structure appeared intact, and the neurological impairments were relatively mild, primarily manifesting as reduced conduction velocity in neurons within the central auditory pathway [14].
CLCN2 mutations have been proved to play a role in familial hyperaldosteronism type 2 [4, 5, 15], and LKPAT [6]. Depienne et al [6] reported six patients with a characteristic pattern of leukoencephalopathy on brain MRI, including three cases presented with adult-onset and three with childhood-onset symptoms. While clinical features overlapped, they showed variability. Adult patients exhibited mild cerebellar ataxia along with differing combinations of chorioretinopathy, visual field abnormalities, optic neuropathy, and headaches. One patient had a schizophrenia-like disorder. The children had mild cerebellar ataxia and a variable combination of mild spasticity, visual field defects, learning disabilities, and headaches. None of the patients had seizures. Brain MRI of all six patients with leukoencephalopathy and ataxia reported by Depienne et al showed prominent signal abnormalities in the middle cerebellar peduncles, midbrain cerebral peduncles, pyramidal tracts in the pons, and posterior limbs of the internal capsule. Depienne et al noted that the clinical features of these patients were nonspecific and did not allow a diagnosis, whereas the MRI findings were specific enough to allow a diagnosis. All six patients were found to have either homozygous or compound heterozygous mutations in the CLCN2 gene [6].
Conclusions
CLCN2-related leukodystrophy is a rare disorder, with characteristic brain MRI findings such as diffuse white matter edema, particularly involving specific tracts within the brainstem. These unique MRI patterns can serve as a strong diagnostic clue for CLCN2 gene analysis. Although our patient did not present with ataxia and therefore could not be considered as a case of LKPAT, interestingly he had characteristic findings of leukodystrophy in his brain MRI, which was then genetically confirmed as a novel CLCN2 mutation. He presented with severe headache and born to consanguineous parents. He was infertile and had mild depression and visual impairment in terms of narrowed visual field. Therefore, we considered him as a case of CLCN2-related leukodystrophy who is, to the best of our knowledge, the oldest case reported in the literature with this mutation.
Acknowledgments
Nothing to declare.
Financial Disclosure
Nothing to declare.
Conflict of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Informed Consent
The patient signed an informed consent form and agreed to the anonymous reporting of his case.
Author Contributions
Slaven Pikija conceived the study, coordinated research activities, and drafted the manuscript. Julia Feige contributed to study design, data collection, analysis, and manuscript revisions. Andreea Toma assisted with data collection, literature review, and analysis. Richard Radlberger provided methodological input and helped refine the manuscript. Eugen Trinka offered clinical insights and contributed to study design and data interpretation. Fritz Klausner performed statistical analyses and assisted with data visualization. Mahdi Safdarian supervised the project and critically revised the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Thiemann A, Grunder S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356(6364):57-60.
doi pubmed - Jentsch TJ, Pusch M. CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev. 2018;98(3):1493-1590.
doi pubmed - Ozaki A, Sasaki M, Hiraide T, Sumitomo N, Takeshita E, Shimizu-Motohashi Y, Ishiyama A, et al. A case of CLCN2-related leukoencephalopathy with bright tree appearance during aseptic meningitis. Brain Dev. 2020;42(6):462-467.
doi pubmed - Fernandes-Rosa FL, Daniil G, Orozco IJ, Goppner C, El Zein R, Jain V, Boulkroun S, et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet. 2018;50(3):355-361.
doi pubmed - Scholl UI, Stolting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, Vichot AA, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet. 2018;50(3):349-354.
doi pubmed - Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, van Berkel C, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 2013;12(7):659-668.
doi pubmed - Guo Z, Lu T, Peng L, Cheng H, Peng F, Li J, Lu Z, et al. CLCN2-related leukoencephalopathy: a case report and review of the literature. BMC Neurol. 2019;19(1):156.
doi pubmed - Sik A, Smith RL, Freund TF. Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience. 2000;101(1):51-65.
doi pubmed - Jeworutzki E, Lopez-Hernandez T, Capdevila-Nortes X, Sirisi S, Bengtsson L, Montolio M, Zifarelli G, et al. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(-) channel auxiliary subunit. Neuron. 2012;73(5):951-961.
doi pubmed - Hoegg-Beiler MB, Sirisi S, Orozco IJ, Ferrer I, Hohensee S, Auberson M, Godde K, et al. Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat Commun. 2014;5:3475.
doi pubmed - Wang H, Xu M, Kong Q, Sun P, Yan F, Tian W, Wang X. Research and progress on ClC-2 (Review). Mol Med Rep. 2017;16(1):11-22.
doi pubmed - Min R, Depienne C, Sedel F, Abbink TEM, van der Knaap MS. CLCN2-related leukoencephalopathy. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews((R)). Seattle (WA). 1993.
pubmed - Bosl MR, Stein V, Hubner C, Zdebik AA, Jordt SE, Mukhopadhyay AK, Davidoff MS, et al. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl(-) channel disruption. EMBO J. 2001;20(6):1289-1299.
doi pubmed - Blanz J, Schweizer M, Auberson M, Maier H, Muenscher A, Hubner CA, Jentsch TJ. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J Neurosci. 2007;27(24):6581-6589.
doi pubmed - Stowasser M, Wolley M, Wu A, Gordon RD, Schewe J, Stolting G, Scholl UI. Pathogenesis of familial hyperaldosteronism type II: new concepts involving anion channels. Curr Hypertens Rep. 2019;21(4):31.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.