| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://jnr.elmerpub.com |
Case Report
Volume 15, Number 3, August 2025, pages 117-122

PIK3CA-Related Overgrowth Spectrum With Progressive Neurological Manifestations: A Rare Case Report
Navina Magesh Kumara, c, Garrett Gianneschib, Janet Elgallabb
aRutgers New Jersey Medical School, Newark, NJ, USA
bDepartment of
Neurology, Rutgers New Jersey Medical School, Newark, NJ, USA
cCorresponding
Author: Navina Magesh Kumar, Rutgers New Jersey Medical School, Newark, NJ, USA
Manuscript submitted June 24, 2025, accepted August 15, 2025, published online August 29,
2025
Short title: A Case of PROS With Neurological Progression
doi:
https://doi.org/10.14740/jnr1036
| Abstract | ▴Top |
PIK3CA-related overgrowth spectrum (PROS) disorders present complex management challenges due to their multisystem involvement. We describe a teenage male with a genetically confirmed PROS disorder (PIK3CA c.353G>A mutation), specifically megalencephaly-capillary malformation-polymicrogyria (MCAP) syndrome, who developed a unique constellation of neurological symptoms. Initially presenting with fetal macrocephaly, seizures, and developmental delay, he underwent surgical interventions including Chiari decompression and ventriculoperitoneal shunt placement. Concurrent progression of scoliosis and gait instability suggested syrinx progression, though obtaining follow-up imaging proved challenging due to insurance barriers. This case uniquely demonstrates the diagnostic complexity of differentiating between primary neurological symptoms, mechanical complications from structural abnormalities, and vascular phenomena in PROS disorders. Management required coordinated care across multiple subspecialties including neurology, neurosurgery, ophthalmology, and orthopedics. The case highlights multiple learning points: 1) the importance of serial monitoring in PROS/MCAP due to the progressive nature of complications, and 2) the challenge of characterizing episodic symptoms in the context of multiple potential underlying mechanisms.
Keywords: PIK3CA-related overgrowth spectrum; Seizures
| Introduction | ▴Top |
PIK3CA-related overgrowth spectrum (PROS) represents a group of rare congenital disorders characterized by tissue overgrowth, with or without cellular dysplasia, in various parts of the body [1]. The condition results from postzygotic gain-of-function variants in PIK3CA that arise during fetal development, causing hyperactivation of the PI3K/AKT/mTOR signaling pathway that ultimately results in tissue overgrowth [1]. Prior to discovery of its shared molecular basis, PROS manifested as distinct but overlapping clinical entities such as megalencephaly-capillary malformation-polymicrogyria (MCAP), congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal syndrome (CLOVES syndrome), hemihyperplasia multiple lipomatosis (HHML), and Klippel-Trenaunay syndrome (KTS) [2].
Specifically, MCAP syndrome is a disorder that encompasses multiple developmental abnormalities including primary megalencephaly (enlarged brain) at birth, vascular (primarily capillary) malformations, distal limb anomalies (e.g. syndactyly), cortical brain malformations (predominantly polymicrogyria), and connective tissue dysplasia. Due to the condition’s evolving nature and risk of complications such as cerebellar tonsillar ectopia, hydrocephalus, and brainstem compression, those with MCAP syndrome require regular monitoring with neuroimaging [2].
| Case Report | ▴Top |
In this case presentation, we present the natural history of XY, a teenage male with genetically verified PROS disorder (c.353G>A mutation) who phenotypically demonstrates characteristics similar to the MCAP syndrome variant of the PROS.
Initial concerns for the health of XY started in utero when fetal macrocephaly was detected on prenatal ultrasound. He was born at 36 weeks via C-section due to macrocephaly and required brief continuous positive airway pressure (CPAP) support. By age 2 months, he developed cutis marmorata telangiectatica. At 3 months old, XY had a first lifetime seizure, characterized by left-sided stiffening, head and eye deviation to the left, and lip smacking. Electroencephalogram (EEG) showed right frontal electrographic seizure, right occipital spikes, and moderate diffuse slowing, and he was started on phenobarbital. Magnetic resonance imaging (MRI) of brain performed at that time showed asymmetric ventricles, with frontal and temporal horns of the right lateral ventricle smaller than the left, and enlargement of the right cerebral hemisphere compared to the left shown in Figure 1. In terms of development, XY had normal gross motor development (stood with support at 11 months, walked with support until about 18 months, walked independently at about 18 months), but had delayed cognitive, social, and speech development with no social smile at 3 months and persistent babbling at 23 months. Given these delays, he was enrolled in early intervention and received physical, occupational, and speech therapy until age 3. Family history is not notable for any neurological disorders, epilepsy, psychiatric disorders, intellectual disabilities, central nervous system (CNS) tumors, or vascular disorders. XY was clinically diagnosed with MCAP within the first year of life. A timeline of events is described below and shown in Figure 2.
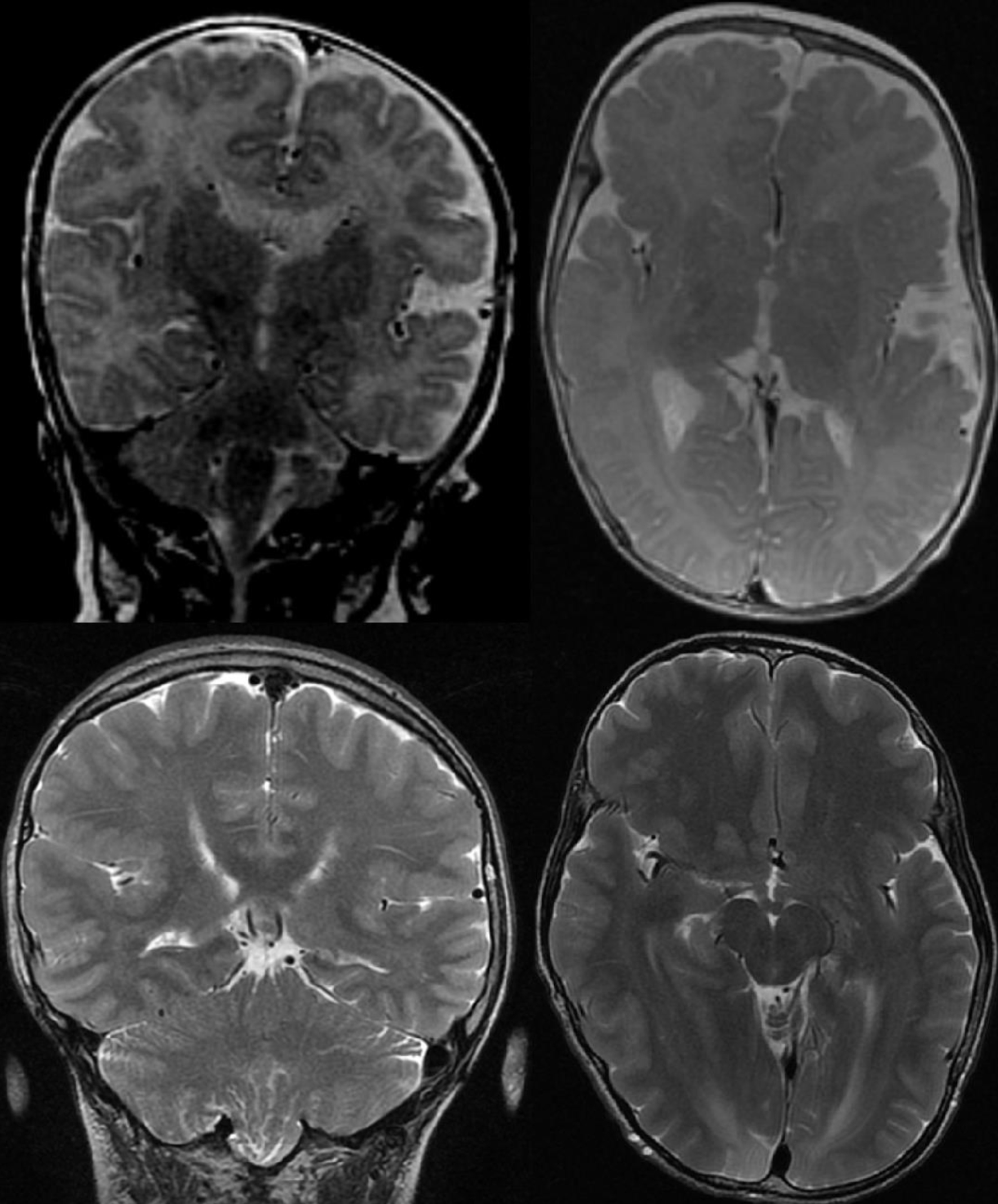 Click for large image |
Figure 1. Top row: brain MRI T2, 2010 (age 3 months). Bottom row: brain MRI T2, 2024 (age 13 years). Note the persistent hemihypertrophy of the right cerebral cortex. MRI: magnetic resonance imaging. |
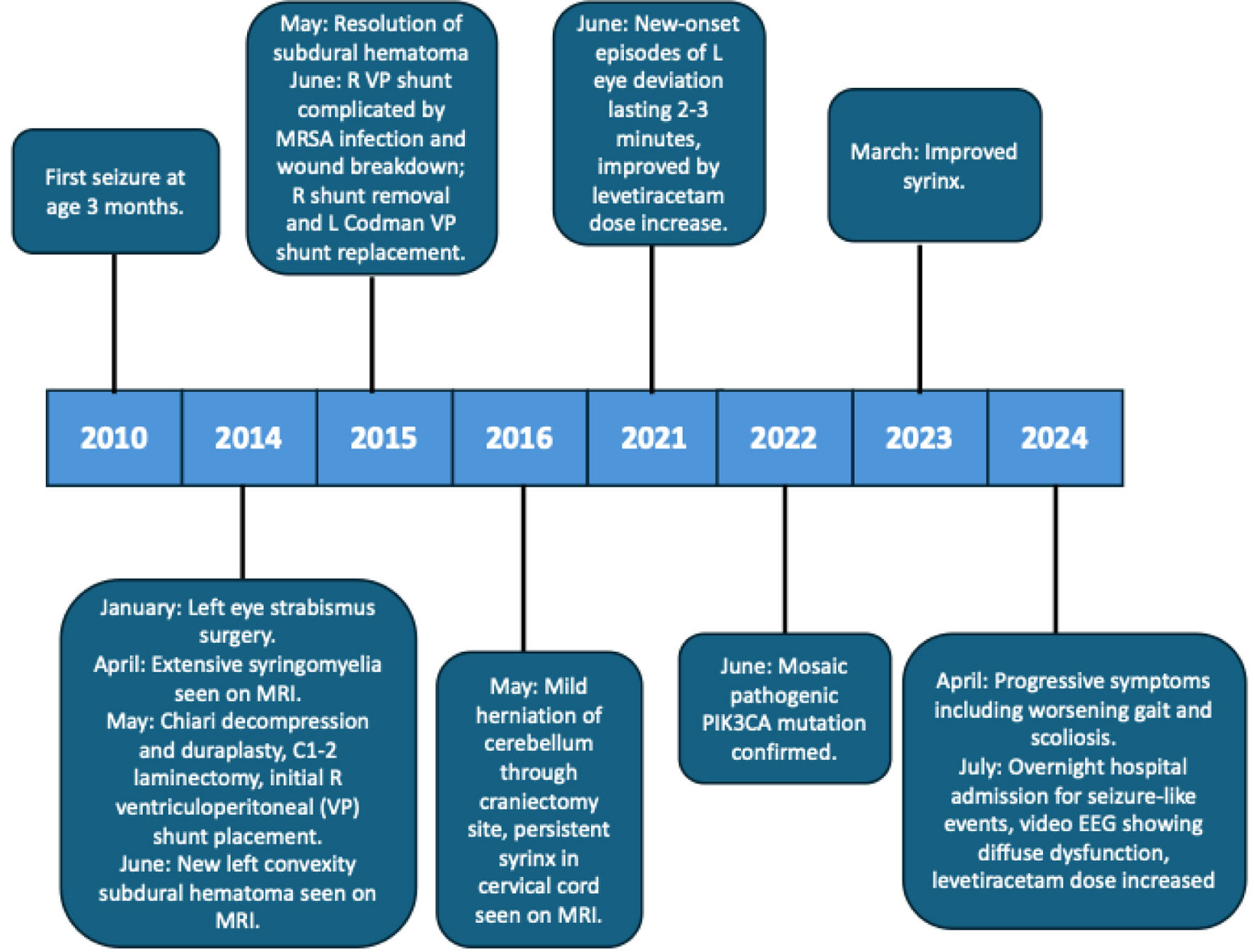 Click for large image |
Figure 2. Timeline. |
Early in infancy XY exhibited distinctive craniofacial features including macrocephaly with frontal and occipital bossing, facial asymmetry with right side enlargement, and micrognathia. Cutaneous manifestations include cutis marmorata telangiectatica and hemangiomas on his hands, left arm, and feet. He also had syndactyly of the left second and third toes which was surgically split and reconstructed with autograft. Due to concern for glaucoma and posterior segment anomalies, which are associated with cutis marmorata telangiectatica congenita, XY was referred to ophthalmology, who continued to follow him for left eye esotropia, which required corrective surgery entailing recession of the left inferior oblique, lateral rectus, and medial rectus muscles, and myopia with astigmatism [3].
At age 2, he required tonsillectomy and adenoidectomy for enlarged tonsils causing obstructive sleep apnea. XY continued to have intermittent seizures with left head and eye deviation and left stiffening. His medication was changed to levetiracetam for concern about phenobarbital’s effect on development. He received genetic testing from an outside hospital, which parents reported to be negative.
At age 3 years, progressive right arm and leg weakness prompted an MRI which showed an extensive syringomyelia extending from C2 to the conus medullaris with a maximum syrinx diameter of 15 mm, likely secondary to a Chiari malformation type 1, shown in Figure 3. The Chiari malformation was decompressed using duraplasty, C1-2 laminectomy and ventriculoperitoneal (VP) shunt placement with revision from the right side to the left side 1 year later after methicillin-resistant Staphylococcus aureus (MRSA) infected the right VP shunt. Despite adequate surgery, the syringomyelia continued to persist to today from C2 to conus medullaris with maximum diameter of 10 mm. Persistence of the syrinx despite decompression could be due to the neuroradiologist-corroborated MRI findings of protrusion of the cerebellum through the craniectomy site and resulting crowding of the neo foramen magnum, or possibly due to dural thickening at the craniectomy site from scar tissue.
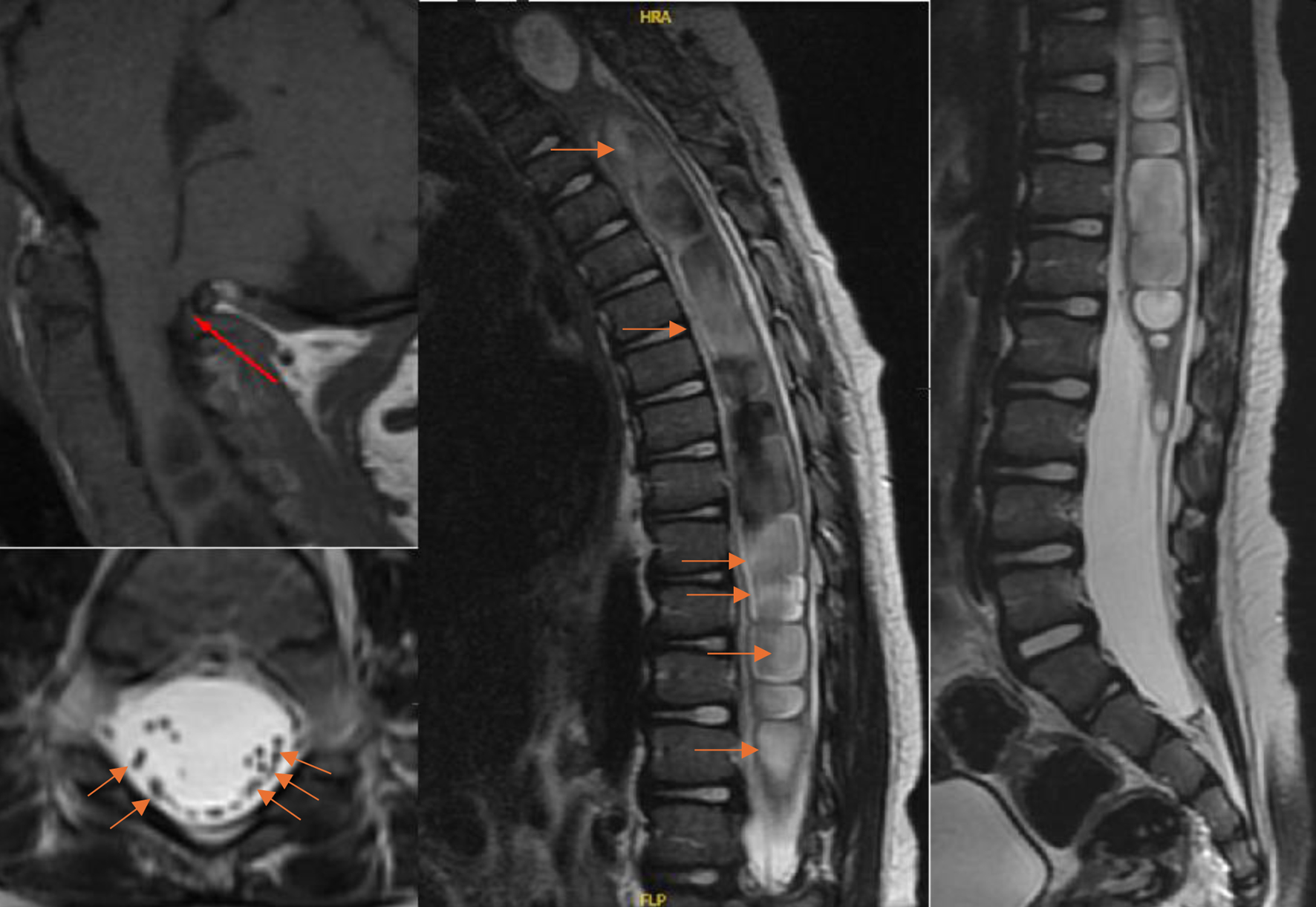 Click for large image |
Figure 3. Top left: MRI cervical spine T1 sagittal view showing Chiari I malformation (red arrow indicating tonsillar herniation shown). Center: MRI T2 thoracic spine sagittal view showing extensive syrinx (red arrows showing fluid appearing hyperintense). Right: MRI T2 lumbar spine sagittal view showing continuation of syrinx. Bottom left: MRI T2 axial view showing scattered cauda equina (red arrows shown). MRI: magnetic resonance imaging. |
Prior to Chiari decompression and VP shunt placement, EEG data showed spikes predominately on the right side, and then after surgery, spikes were bilateral predominating on the left side, as shown in Figure 4. Seizure semiology also changed from left head and eye deviation to generalized tonic-clonic episodes. Seizures were well controlled on levetiracetam monotherapy 1,800 mg twice a day (75 mg/kg/day); however, two seizures in the past 10 years were both generalized tonic-clonic. The left-sided spike predominance on EEG has also continued into teenage years. Of note, intermittent high amplitude 20 - 22 Hz localized fast activities secondary to dysplastic cortex have persisted into teenage years as well.
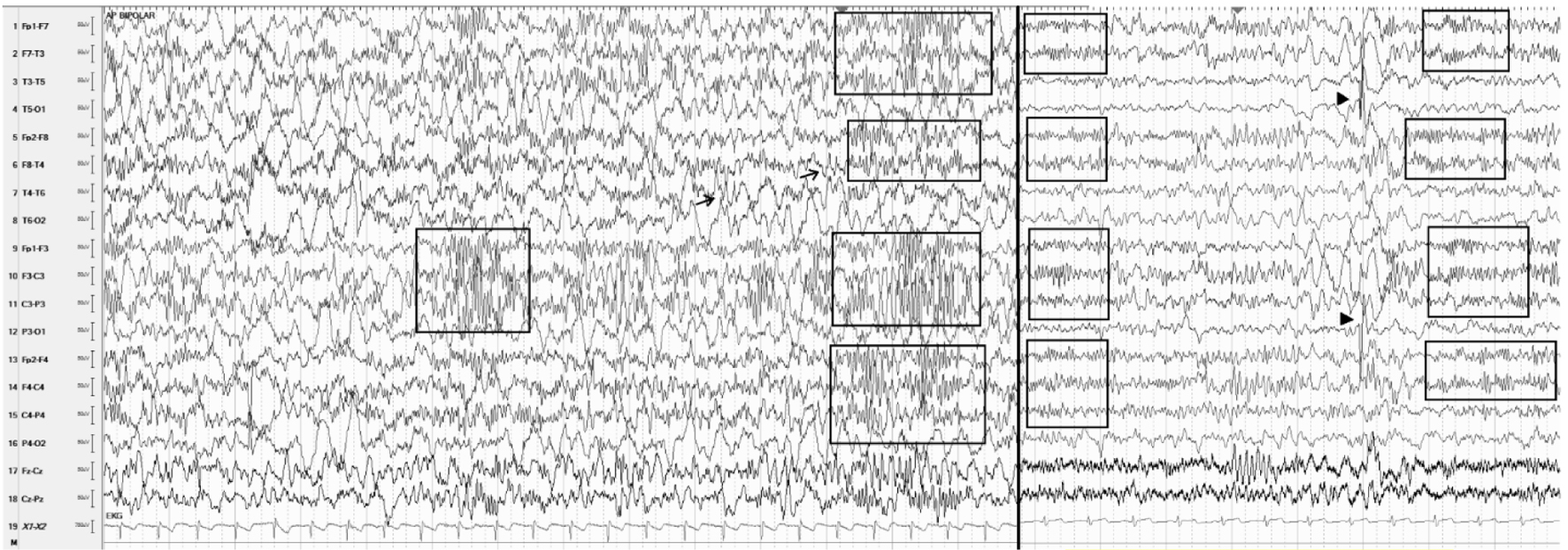 Click for large image |
Figure 4. Representative samples of EEGs. Left: at 3 years old while on levetiracetam, prior to surgery sensitivity 7 µV/mm excessive background slowing with spikes at right temporal area (arrows) and intermittent 20 - 22 Hz bilateral multifocal fast activity (boxes). Right: EEG at age 12, sensitivity 15 µV/mm with persistent fast activity (boxes) and T5, O1, P3 spikes (arrowheads) while on levetiracetam. EEG: electroencephalogram. |
Genetic confirmation of c.353G>A mutation in PIK3CA gene was performed via next-generation sequencing when XY was 12 years old. Albeit rare, this variant has been identified in another case of PROS, malignant tumors, Cowden disease, and angio-osteo-hypertrophic syndrome [4].
Considering XY’s many neurological complications, he has received regular neuroimaging throughout his life. After the Chiari decompressive surgery and VP shunt revision, MRI results of the brain and spine have been relatively stable into teenage years. He continues to have right cerebral hemisphere overgrowth, enlargement of the cerebellum, diffuse polymicrogyria, holocord syrinx, and Chiari malformation I with foramen magnum crowding.
Currently, many of the dysmorphic, musculoskeletal, and cutaneous features have persisted into teenage life including facial asymmetry with right side enlargement, micrognathia, cutis marmorata telangiectatica and hemangiomas on his hands, left arm, and feet, macrocephaly with frontal and occipital bossing with hemimegalencephaly, shown in Figure 1. Musculoskeletal examination reveals worsening thoracolumbar scoliosis (Cobb angle 23° from T7-L1), hypertrophy of right thigh, < 1st percentile for height, and a right leg that is longer than the left leg by 2 cm. His current physical exam demonstrates unsteady gait requiring multiple corrective steps, bilateral brisk reflexes with clonus, downgoing plantar reflexes, 4/5 strength in bilateral ankle dorsiflexion and plantar flexion, 4/5 strength in left hip flexion, no visual field cuts, left gaze preference, and left eye nystagmus with right gaze. The left eye nystagmus has only been noted in more recent years and is unclear in etiology, although it is worth noting that the patient has cerebellar enlargement and underwent surgery for the same eye during early childhood. His development is marked by intellectual disability with preserved social interaction and intact, but slow speech in both English and his native language. He is currently in seventh grade in special education classes and receives services from speech-language pathology, physical therapy, and occupational therapy in school as per an Individualized Education Program. The patient continues to receive coordinated care across multiple subspecialities including neurology, neurosurgery, ophthalmology, and orthopedics.
| Discussion | ▴Top |
This case highlights several key aspects of PROS/MCAP syndrome management. The progressive nature of neurological manifestations, from initial presentation with macrocephaly and seizures to the development of nystagmus, motor weakness, and gait instability, emphasizes the importance and effectiveness of longitudinal monitoring.
From the neurology perspective, our team’s approach demonstrated thorough diagnostic evaluation. The early recruitment and maintenance of coordinated multidisciplinary care allowed for comprehensive management of multiple organ system involvement and arrangement of supportive management outside the clinical setting such as physical therapy, occupational therapy, and speech therapy. Regular follow-up allowed for timely adjustment of antiepileptic medications. More recently, however, regular imaging surveillance was hampered by insurance barriers, potentially delaying recognition of disease progression and the need for surgical intervention. This obstacle posed by insurance is particularly bothersome as there is international consensus that yearly MRI scans are necessary for those diagnosed with CNS overgrowth or hyperplasia at a young age to monitor for progressive ventriculomegaly and cerebellar tonsillar herniation [5, 6].
In this case report, we observed persistent, bilateral, intermittent high-frequency beta rhythm across a decade of EEG monitoring. Seizures have been well documented in 30-40% of PIK3CA syndrome cases; however, background activity has not been well documented [1]. High frequency beta is typically a marker for polymicrogyria and other malformations of cortical development. Our patient has bilateral multifocal polymicrogyria and bilateral multifocal high frequency beta, suggesting that this abnormal rhythm is localized to the abnormal cortical structures. One prior report described drug-refractory epilepsy in a patient with somatic mosaicism for a PIK3CA mutation localized to a malformation of cortical development in the left frontal lobe, showing frequent spikes and repetitive fast activity on stereo-EEG depth recordings [7].
In a brief review of the literature, seizure foci are typically unilateral and ipsilateral to the hemimegalencephaly, likely corresponding to the malformations of cortical development secondary to the hemimegalencephaly [8]. Also, MCAP and PIK3CA syndromes often have bilateral malformations of cortical development, such as the bilateral polymicrogyria seen in our case [9-11]. Cortical dysplasia in general tends to act as seizure foci [1]. Therefore, there appears to be an added/extra seizure tendency produced by the hemimegalencephalic tissue over and above the baseline seizure tendency produced by malformations of cortical development. Of note, our case showed early seizure foci localizing to the hemimegalencephalic side which evolved during late childhood into bilateral, multifocal seizure foci producing generalized tonic-clonic seizures. Since seizure types and abnormal EEG findings largely depend on the tissue distribution of pathogenic PIK3CA variants, perhaps polymicrogyria becomes the more prominent cortical anomaly compared to megalencephaly given that the increased growth rate at birth slows to a normal rate within the first few years of life. This evolution of focal to generalized seizures appears to represent a unique finding as previously documented cases of MCAP syndrome tend to involve focal seizures [2, 12]. Moreover, although PIK3CA pathogenic variants are linked to epilepsy, current reviews of PROS disorders do not differentiate between focal and generalized seizures nor delineate the epileptic foci per EEG [1].
This case underscores the complexity of PROS disorders and highlights the challenges in managing progressive, rare genetic disorders within the current healthcare system. However, there lies hope for more precise treatment beyond supportive management, with new research studies involving targeted therapy underway [13, 14]. For instance, in a study evaluating BYL719, a PIK3CA inhibitor, all 17 patients with PROS secondary to 10 distinct gain-of-function mutations demonstrated clinical improvement. This included pediatric and adult patients (mean age 14.5 years, SD 11.4, range 4 - 50). Among the 17 patients, two had been diagnosed with MCAP and both experienced improved cognition and behavior, possibly explained by an improvement in cerebral perfusion. Furthermore, one experienced a reduction in hemifacial hypertrophy whereas the other experienced improvement of scoliosis and improvement of chronic, steroid-dependent cellulitis [13]. As of 2022, alpelisib is an FDA-approved PI3Ka-specific inhibitor for patients with PROS, although during its development, it was unclear whether alpelisib was efficacious in improving neurocognitive functioning in patients with MCAP syndrome as it is uncertain whether the drug crosses the blood-brain barrier. The SESAM trial, a two-period multicenter French phase II trial, will evaluate efficacy of alpelisib in MCAP patients via a primary outcome of ≥ 4-point improvement in the Vineland II Adaptive Behaviour Scale (VABS) following 24 months of treatment [14]. Although early experience with PI3K inhibitors suggests clinical promise, definitive evidence in MCAP remains limited, particularly regarding neurocognitive outcomes, CNS penetration and pharmacodynamics, optimal timing and dosing in mosaic disease, durability of response, and long-term safety. Accordingly, longitudinal genotype- and phenotype-stratified studies with standardized endpoints are needed to establish efficacy thresholds and shape future evidence-based guidelines concerning care of MCAP patients.
Acknowledgments
We are grateful to the patient and family for permitting long-term clinical care and documentation.
Financial Disclosure
The authors received no financial support for the research, authorship, or publication of this article.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
This case has been de-identified. Given the retrospective nature and lack of identifiable data, informed consent was not required.
Author Contributions
Navina Magesh Kumar, BS: primary author, neurological data synthesis and manuscript drafting. Garrett Gianneschi, MD: radiologic interpretation, clinical data compilation, and manuscript drafting. Janet Elgallab, MD: supervision, manuscript editing, and critical review.
Data Availability
The authors declare that all relevant clinical data supporting the findings of this study are available within the article are included in the manuscript. Additional information can be made available upon request in accordance with institutional policy and patient confidentiality.
| References | ▴Top |
- Mirzaa G, Graham JM, Jr., Keppler-Noreuil K. PIK3CA-related
overgrowth spectrum. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds.
GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. 1993.
pubmed - Sarma K, Nayak MK, Mishra B, Gaikwad SB. Megalencephaly-capillary
malformation-polymicrogyria syndrome (MCAP): a rare dynamic genetic disorder. Cureus.
2022;14(5):e25123.
doi pubmed - Elitt MS, Tamburro JE, Moran RT, Traboulsi E. Cutis marmorata
telangiectatica congenita: a focus on its diagnosis, ophthalmic anomalies, and possible
etiologic factors. Ophthalmic Genet. 2020;41(2):101-107.
doi pubmed - Ahakoud M, Daha Belghiti H, Ihlal H, Bouguenouch L. A mosaic PIK3CA
mutation in a moroccan female: exploring the diagnostic challenges of PIK3CA-related overgrowth
spectrum. Cureus. 2023;15(4):e36996.
doi pubmed - Mirzaa GM, Conway RL, Gripp KW, Lerman-Sagie T, Siegel DH, deVries
LS, Lev D, et al. Megalencephaly-capillary malformation (MCAP) and
megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related
disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med
Genet A. 2012;158A(2):269-291.
doi pubmed - Douzgou S, Rawson M, Baselga E, Danielpour M, Faivre L, Kashanian A,
Keppler-Noreuil KM, et al. A standard of care for individuals with PIK3CA-related disorders: An
international expert consensus statement. Clin Genet. 2022;101(1):32-47.
doi pubmed - Phillips HW, D'Gama AM, Wang Y, Chahine Y, Chiu M, Swanson AC, Ahtam
B, et al. Somatic mosaicism in PIK3CA variant correlates with
stereoelectroencephalography-derived electrophysiology. Neurol Genet.
2024;10(1):e200117.
doi pubmed - Dobyns WB, Mirzaa GM. Megalencephaly syndromes associated with
mutations of core components of the PI3K-AKT-MTOR pathway: PIK3CA, PIK3R2, AKT3, and MTOR.
Am J Med Genet C Semin Med Genet. 2019;181(4):582-590.
doi pubmed - Park HJ, Shin CH, Yoo WJ, Cho TJ, Kim MJ, Seong MW, Park SS, et al.
Detailed analysis of phenotypes and genotypes in megalencephaly-capillary
malformation-polymicrogyria syndrome caused by somatic mosaicism of PIK3CA mutations.
Orphanet J Rare Dis. 2020;15(1):205.
doi pubmed - de Kock L, Cuillerier A, Gillespie M, Couse M, Hartley T, Mears W,
Bernier FP, et al. Molecular characterization of 13 patients with PIK3CA-related overgrowth
spectrum using a targeted deep sequencing approach. Am J Med Genet A.
2024;194(3):e63466.
doi pubmed - Garde A, Guibaud L, Goldenberg A, Petit F, Dard R, Roume J,
Mazereeuw-Hautier J, et al. Clinical and neuroimaging findings in 33 patients with MCAP
syndrome: A survey to evaluate relevant endpoints for future clinical trials. Clin Genet.
2021;99(5):650-661.
doi pubmed - Segal D, Heary RF, Sabharwal S, Barry MT, Ming X. Severe holocord
syrinx in a child with megalencephaly-capillary malformation syndrome. J Neurosurg
Pediatr. 2016;18(1):79-82.
doi pubmed - Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, Blanc E,
et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature.
2018;558(7711):540-546.
doi pubmed - Luu M, Vabres P, Espitalier A, Maurer A, Garde A, Racine C,
Carpentier M, et al. A phase II double-blind multicentre, placebo-controlled trial to assess the
efficacy and safety of alpelisib (BYL719) in paediatric and adult patients with
Megalencephaly-CApillary malformation Polymicrogyria syndrome (MCAP): the SESAM study protocol.
BMJ Open. 2024;14(12):e084614.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Neurology Research is published by Elmer Press Inc.