| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://jnr.elmerpub.com |
Case Report
Volume 15, Number 4, December 2025, pages 203-210

Multifocal Bihemispheric Ischemic Strokes Due to Unknown Thrombotic Microangiopathy
Taseal Ahmeda, d, Yiorgos Antoniadisa, Genesis Reyes Vegab, Samir Ruxmohanc
aSt. George’s University School of Medicine, True Blue,
Grenada
bUniversity of Medicine and Health Sciences, Basseterre, Saint Kitts and
Nevis
cExceed Healthcare, Dallas, TX 75208, USA
dCorresponding
Author: Taseal Ahmed, St. George’s University School of Medicine, True Blue, Grenada
Manuscript submitted June 30, 2025, accepted September 24, 2025, published online December 24,
2025
Short title: Ischemic Stroke in Thrombotic Microangiopathy
doi:
https://doi.org/10.14740/jnr1040
| Abstract | ▴Top |
Thrombotic microangiopathy is an uncommon etiology of acute ischemic stroke, particularly when accompanied by overlapping features of multiple subtypes of thrombotic microangiopathies. Our case highlights the diagnostic and therapeutic challenges that emerge when stroke, hemolysis, coagulopathy, and renal dysfunction converge in a single clinical scenario. We present a 63-year-old female that developed multifocal, bilateral cerebral infarctions in the context of severe thrombocytopenia, hemolytic anemia, prolongation of clotting times, and acute kidney injury. The trajectory of her laboratory parameters revealed evolving coagulopathy and kidney injury, hallmarks of both thrombotic thrombocytopenic purpura and disseminated intravascular coagulation. In addition, normal ADAMTS13 activity level led to diagnostic uncertainty and delay in initiating plasma exchange and immunosuppression. The patient’s clinical course rapidly deteriorated, resulting in fatal neurologic injury. The patient’s course reveals the challenges of distinguishing among thrombotic microangiopathies when clinical and laboratory findings are incomplete. This ambiguity delayed initiation of plasma exchange and corticosteroids, and neurological outcomes were ultimately terminal. Our experience exemplifies key clinical principles: a normalized ADAMTS13 result obtained after plasma transfusion should not exclude thrombotic thrombocytopenic purpura; and, when suspicion remains high, empiric plasma exchange and immunosuppression should be instituted without delay, as survival depends on rapid intervention. Conversion of laboratory and clinical data into timely action remains essential to improving outcomes in such complex presentations. This case illustrates a unique presentation of multifocal ischemic stroke due to an evolving thrombotic microangiopathy with disseminated intravascular coagulation-like features, emphasizing the need for heightened clinical vigilance and decisive treatment.
Keywords: Thrombotic microangiopathy; Disseminated intravascular coagulation; Thrombotic thrombocytopenic purpura; ADAMTS13; Multifocal ischemic strokes; Plasma exchange
| Introduction | ▴Top |
Microangiopathic hemolytic anemias (MAHAs) are hemolytic disorders defined by intravascular fragmentation of red blood cells (RBCs) within the microcirculation, leading to characteristic schistocytes on peripheral blood smear and laboratory evidence of hemolysis (elevated lactate dehydrogenase, indirect bilirubin, and low haptoglobin) [1]. This phenomenon usually signifies an underlying thrombotic microangiopathy (TMA), defined as a syndrome of thrombocytopenia, MAHA, and small-vessel thrombosis causing end-organ ischemia [2]. Primary TMAs include thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). TTP results from severe ADAMTS13 metalloprotease deficiency (often immune-mediated by anti-ADAMTS13 autoantibodies, or rarely inherited via ADAMTS13 gene mutations), while HUS may be “typical” (Shiga toxin-mediated) or “atypical” due to dysregulated complement activity [3]. Secondary TMAs arise in background of systemic clinical contexts, for example, pregnancy-related syndromes (e.g., HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome, preeclampsia/eclampsia), malignant hypertension, autoimmune diseases like systemic lupus erythematosus or antiphospholipid syndrome (APS), certain drug toxicities (e.g., quinine, calcineurin inhibitors, chemotherapies, antiplatelet agents), severe infections (such as human immunodeficiency virus (HIV) or sepsis), malignancies, and post-transplant settings [2]. Consideration also be given to coagulopathy-associated microangiopathy like disseminated intravascular coagulation (DIC), a consumptive coagulopathy triggered by causes like severe infection, trauma, malignancy or obstetric complications [3]. DIC shares the features of microvascular thrombosis and MAHA seen in TMA but is distinguished by laboratory evidence of widespread coagulation activation and fibrinolysis (e.g., prolonged clotting times, elevated fibrin degradation products), reflecting a global coagulopathic process rather than an isolated platelet-von Willebrand factor (vWF) microthrombi process [3].
Regardless of the underlying etiology, TMAs can precipitate acute ischemic stroke via microvascular platelet-rich thrombi that occlude small cerebral arterioles and capillaries, producing multifocal areas of ischemia even in the presence of thrombocytopenia [2]. Endothelial injury and ensuing microthrombosis in the cerebral circulation are especially well-recognized in TTP and in complement-mediated HUS (atypical HUS), but similar neurologic ischemic complications have also been documented in secondary TMA contexts such as pregnancy-associated TMAs, malignant hypertension, and DIC. The brain’s high metabolic demand and dense microvascular network render it particularly vulnerable to ischemic injury in these settings, making timely recognition and targeted treatment critical to prevent permanent neurologic deficits [2]. Coagulation disorders as a whole account for < 5% of ischemic stroke, and TMA-associated strokes are uncommon but carry high risk of mortality [1]. Ischemic stroke has been described most often in the context of TTP, yet even in TTP it occurs in only a minority of cases (roughly 8-10% of TTP hospitalizations involve an acute ischemic stroke) [1]. Despite their low incidence rate, strokes due to TMA are crucial to recognize because TMAs are life-threatening if untreated and require prompt, specific therapy [1]. In clinical practice, any unexplained stroke occurring alongside hemolytic anemia and thrombocytopenia should prompt immediate concern for an underlying TMA process, as timely plasma exchange (PLEX) or other targeted treatment can be lifesaving in these patients [1]. The diagnostic overlap of TMA presentations often mimic more common stroke etiologies, and classic TMA laboratory findings may be incomplete or confounded by other factors. A high index of suspicion must be maintained even if definitive hematological markers or coagulation studies have not returned yet or are within reference ranges. In the following case, we describe a patient with multifocal ischemic strokes in the setting of a suspected TMA with overlapping laboratory TMA subtype features, highlighting the clinical consequences of diagnostic uncertainty.
| Case Report | ▴Top |
A 63-year-old female with a past medical history of coronary artery disease status post percutaneous coronary intervention to right coronary artery, ischemic cardiomyopathy with ejection fraction of 25%, chronic deep vein thrombosis (DVT) on apixaban with an inferior vena cava filter, multiple past cerebrovascular accidents and previous history of polysubstance abuse was transferred to our center for evaluation of severe anemia and hypotensive shock requiring vasopressor support. Initially, she presented to an outside hospital with profound anemia (hemoglobin 5 g/dL, reference 12.0 - 16.0 g/dL) in the setting of suspected new onset cirrhosis. Gastrointestinal bleeding was suspected, but no source was identified, and hepatology evaluation determined no cirrhosis. She received urgent transfusions of one unit of packed RBCs, two units of plasma, and two units of cryoprecipitate for stabilization. Despite hemodynamic resuscitation, the patient remained encephalopathic, with intermittent unresponsiveness and disorientation. On day 1 of admission, her mental status worsened acutely. She became nonverbal, stopped following commands, stopped visually tracking, and developed right hemiplegia. Neurology consultation was obtained for concern of stroke. On initial neurological examination, her National Institutes of Health Stroke Scale (NIHss) score was 19, indicating a severe stroke syndrome with global aphasia, forced leftward gaze deviation, and right-sided flaccid paralysis. An initial magnetic resonance imaging (MRI) of the brain demonstrated recent ischemic strokes in bilateral cerebral hemispheres, including evolving infarcts in the left temporal and occipital lobes (from prior MRI imaging) and new diffusion-restricted lesions in the right frontal and parietal lobes, without hemorrhage or mass effect (Fig. 1). These acute infarcts coexisted with chronic microvascular ischemic changes and old encephalomalacic infarcts in the bilateral occipital and right parietal lobes (Fig. 1). There were no signs of active infection on urinalysis, and urine toxicology screen was negative. Duplex ultrasonography of the legs showed no DVT to explain an embolic source. Given her anticoagulated status and subacute presentation, thrombolytic therapy was not pursued in addition to not meeting standard time criteria.
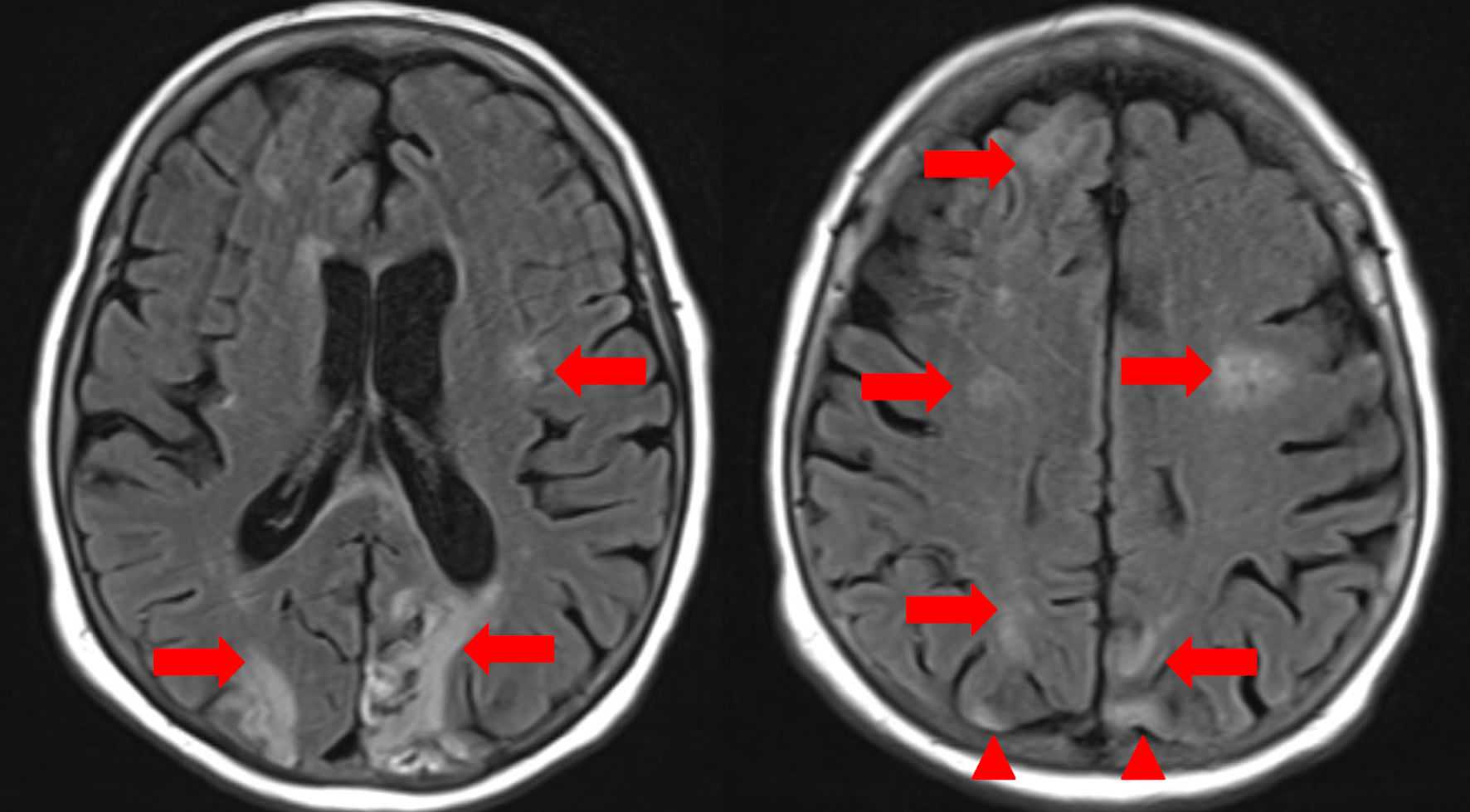 Click for large image |
Figure 1. Magnetic resonance imaging (MRI) of the brain, axial T2-weighted fluid-attenuated inversion recovery (FLAIR) sequences. Red arrows indicate areas of persistent hyperintensity consistent with evolving acute ischemic infarcts in the bilateral cerebral hemispheres, notably involving the frontal, parietal, and periventricular white matter regions. Additional foci in the right frontal and parietal lobes suggest new acute infarcts. Confluent periventricular hyperintensities are present, compatible with moderate chronic microvascular ischemic disease. Red arrowheads highlight areas of subacute-to-chronic ischemic injury/encephalomalacia, demonstrated by parenchymal volume loss and gliotic signal in the bilateral occipital lobes and right parietal lobe. Multiple small chronic lacunar infarcts are also seen in the corona radiata and centrum semiovale. |
On hospital day 2, the patient’s neurological status deteriorated further, necessitating endotracheal intubation for airway protection. Vascular imaging by computed tomography (CT) angiography showed an occlusion of the right internal carotid artery (ICA) at its origin, with collateral flow maintaining patency of the distal ICA via the circle of Willis. The right middle cerebral artery (MCA) and anterior cerebral artery had markedly diminished flow in the right hemisphere. Emergent mechanical thrombectomy was performed for the right ICA occlusion approximately 18 h after her initial stroke onset. During endovascular therapy, there was a small perforation in a right MCA branch with contrast extravasation, managed by tamponade with the catheter, and active bleeding ceased after 30 min of low-pressure inflation. The intervention achieved successful reperfusion of the right anterior circulation (Thrombolysis in Cerebral Infarction grade 3), extracting a large volume thrombus from the carotid terminus. Post-procedure, however, the patient remained deeply comatose (Glasgow Coma Scale 5T) with absent brainstem reflexes (non-reactive pupils, unilateral loss of corneal reflex) and flaccid quadriparesis. Her extremities were mottled with dusky purple discoloration and weak peripheral pulses, raising concern for systemic hypoperfusion or microvascular thrombi. Electroencephalography showed diffuse slowing without epileptiform activity, consistent with severe diffuse cerebral dysfunction. Complete blood counts after thrombectomy showed persistent anemia (hemoglobin 5 g/dL, reference 12.0 - 16.0 g/dL) with normal mean corpuscular volume (MCV, 80.1, reference 80 - 100) and marked thrombocytopenia (platelet count 44 × 103/µL, reference 130 - 400 × 103/µL). A peripheral blood smear demonstrated schistocytes consistent with microangiopathic hemolysis. Hemolysis labs were notable for indirect hyperbilirubinemia (indirect bilirubin 3.1 mg/dL, reference < 1.0 mg/dL) and elevated lactate dehydrogenase (LDH 1,223 U/L, reference 100 - 250 U/L), suggesting ongoing hemolytic anemia. Coagulation studies revealed features of consumptive coagulopathy: fibrinogen was critically low (< 60 mg/dL, reference 214 - 481 mg/dL) and prothrombin time (PT)/international normalized ratio (INR) was prolonged (PT/INR 34.0/3.6, reference 11.3 - 14.7/0.9 - 1.2) (see Table 1 for the temporal evolution of hematologic and coagulation parameters following thrombectomy). Her blood urea nitrogen elevated from 43 to 56 (reference 10 - 25) and creatinine was elevated from 2.2 to 2.9 (reference 0.7 - 1.4), indicating acute kidney injury. She received transfusions of two units of leukoreduced packed RBCs, two units of fresh frozen plasma, and one unit of cryoprecipitate. Hematology was urgently consulted for strong concern for a TMA such as TTP, atypical HUS, DIC, or catastrophic antiphospholipid syndrome (CAPS). Notably, an ADAMTS13 activity assay (drawn after initial plasma transfusions at the outside hospital) returned at 55% of normal, not diagnostic for TTP but potentially falsely elevated by the initial plasma infusion. Neurology recommended empirically covering TTP in the differential with high-dose corticosteroids and PLEX, but these therapies were initially deferred by hematology due to concerns about the patient’s tenuous condition (worsening renal failure and coagulopathy) and the unclear etiology of the strokes. Anticoagulation (for a possible APS) was also held due to the high hemorrhagic risk in the setting of large strokes and coagulopathy.
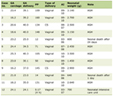 Click to view |
Table 1. Hematologic, Biochemical, and
Coagulation Parameters After Thrombectomy |
Laboratory values revealed worsening anemia, persistent thrombocytopenia, and rising markers of renal dysfunction. These trends are summarized in Table 1, which highlights the evolving coagulopathy and renal dysfunction. Coagulation studies demonstrated critically low fibrinogen, prolonged INR, and indirect hyperbilirubinemia, further blurring the distinction between TTP, complement-mediated TMA, and DIC.
By hospital day 3, follow-up head CT demonstrated extensive right-hemispheric swelling with a progressive midline shift (7 mm to 12 mm), evolving acute ischemia in the posterior and anterolateral right frontal lobes, and diffuse cerebral edema consistent with evolving infarction, without hydrocephalus (Fig. 2). Repeat imaging with dual energy CT showed contrast in the subarachnoid space with no hemorrhage but revealed midline shift (12 mm) with progressive infarction in the right frontal and parietal lobes (Fig. 3). There was no acute hydrocephalus, but there were radiographic signs of increased intracranial pressure (ICP) (Fig. 2). Lumbar puncture was contraindicated by her coagulopathy and imaging findings. Laboratory testing showed hemoglobin 6.7 g/dL (reference 12.0 - 16.0) and hematocrit 21.6% (reference 36.0 - 46.0). Two units of cryoprecipitate were administered and post-transfusion labs showed that hemoglobin had risen to 7.3 g/dL (reference 12.0 - 16.0) and hematocrit to 23.4% (reference 36.0 - 46.0), with persistent thrombocytopenia (platelets 58 × 103/µL, reference 130 - 400) and normocytic indices (MCV 81.5 fL, reference 80.0 - 100.0). Chemistry and coagulation studies demonstrated hyperbilirubinemia (total bilirubin 4.7 mg/dL; indirect bilirubin 3.4), azotemia (blood urea nitrogen (BUN) 62 mg/dL, reference 10 - 25, creatinine 3.50 mg/dL, reference 0.70 - 1.40), and coagulopathy (prothrombin time 26.4 s reference 11.3 - 14.7, INR 2.6 reference 0.9 - 1.2). The patient’s clinical status continued to deteriorate despite maximal supportive measures, including hyperosmolar therapy (mannitol) for cerebral edema and strict blood pressure control to minimize further stroke extension. Broad rheumatologic and infectious workups (antinuclear antibody (ANA), antineutrophil cytoplasmic antibodies (ANCA), rheumatoid factor, anti-cyclic citrullinated peptide antibodies, antiphospholipid antibodies, vasculitis panel, rapid plasma reagin (RPR) for syphilis) were unremarkable. Therapeutic PLEX and methylprednisolone were initiated for TTP as it remained a leading possibility (given thrombocytopenia with microangiopathic hemolysis). Despite maximal supportive measures including ICP-directed therapy, ventilatory management, and PLEX, the patient’s neurological status did not improve. Given the extent of infarction, lack of brainstem reflexes, and poor prognosis, multidisciplinary goals-of-care discussions were held with the family. In light of irreversible neurologic injury and systemic deterioration, the decision was made to transition to comfort-focused care with withdrawal of life-sustaining therapies.
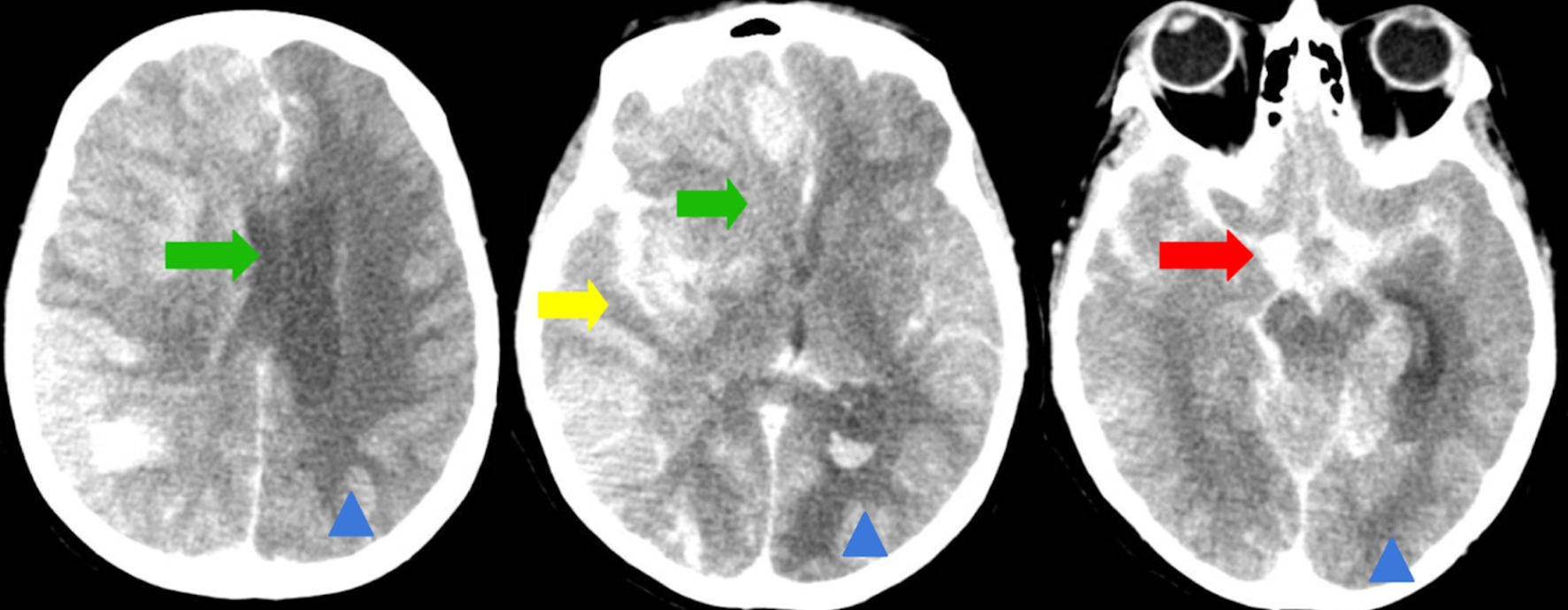 Click for large image |
Figure 2. Computed tomography (CT) of the brain (axial non-contrast). Green arrows indicate a worsening leftward midline shift, now measuring approximately 12 mm (previously 7 - 8 mm), due to significant hemispheric mass effect. Yellow arrow highlights hypodensity and swelling in the brainstem and cerebellum, consistent with acute cerebral edema. Red arrow denotes acute subarachnoid hemorrhage within the basal cisterns and right-sided cerebral sulci. Chronic ischemic changes, characterized by volume loss and gliosis in the right occipital lobe and left cerebral hemisphere, would be marked with blue arrowheads for distinction from acute pathology. |
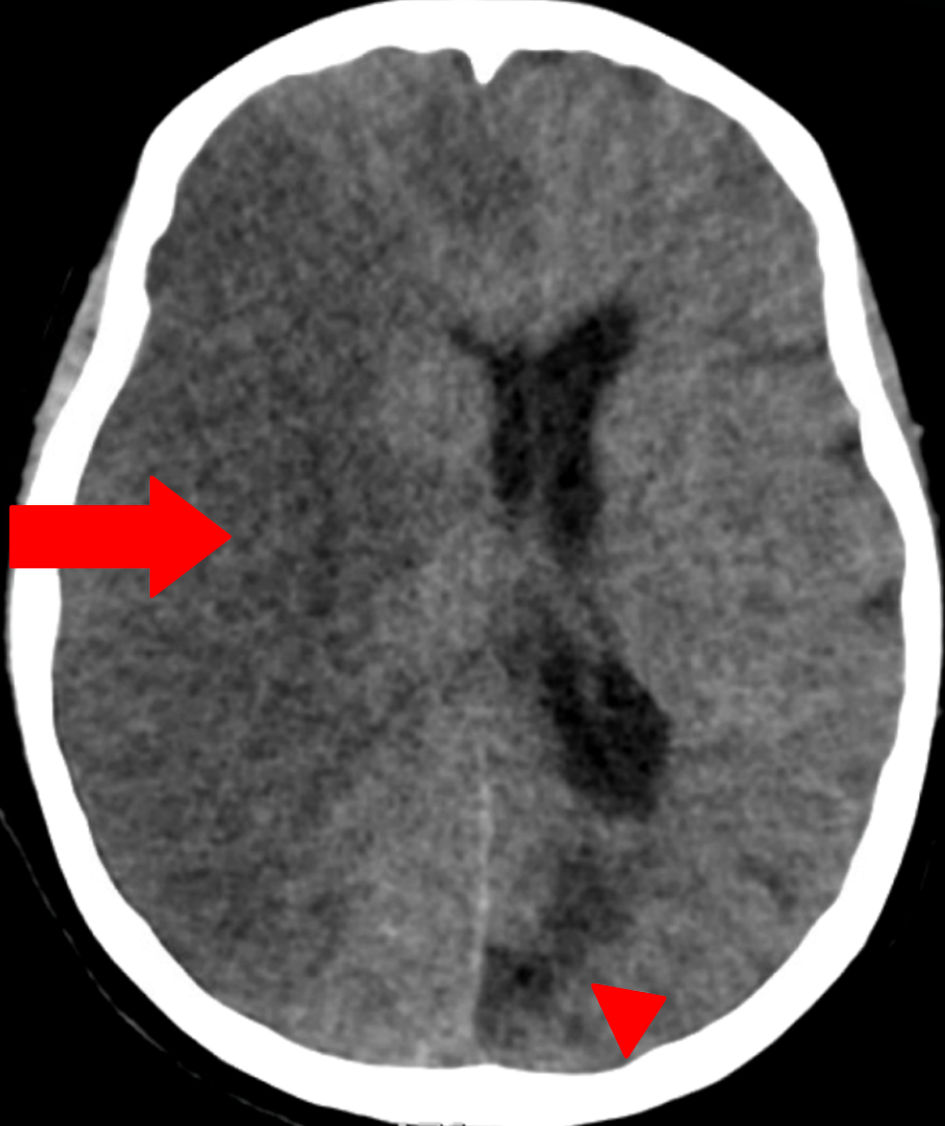 Click for large image |
Figure 3. Dual-energy computed tomography (DECT) of the head (axial view). Red arrow indicates a large area of hypodensity involving the right cerebral hemisphere, associated with pronounced mass effect and midline shift, consistent with an extensive acute-to-subacute ischemic infarct. No subarachnoid hemorrhage is seen. Red arrowhead marks an area of chronic ischemic injury/encephalomalacia in the left occipital region, characterized by parenchymal volume loss and gliosis. The use of arrowheads differentiates chronic lesions from acute/subacute pathology. |
| Discussion | ▴Top |
This case highlights the importance of a systematic diagnostic approach when ischemic strokes occur with unusual features including multi-territorial distribution, concurrent coagulopathy, and end-organ injury. While common stroke mechanisms (atherothrombosis or cardioembolism) were considered, the bilateral and diffuse nature of infarcts alongside laboratory evidence of hemolysis and coagulopathy pointed toward a TMA as the underlying etiology. Coagulation disorders are an uncommon cause of ischemic stroke (< 5% of cases) but should be considered on the differentials list in the right clinical context [1]. Thrombotic microangiopathies encompass entities like TTP, HUS (including atypical HUS), DIC, and CAPS, among others [3]. All TMAs share a common pathogenesis of microvascular thrombosis causing organ ischemia, thrombocytopenia from platelet consumption, and MAHA from red cell fragmentation [1]. In our patient, the combination of thrombocytopenia, schistocytes, elevated indirect bilirubin, acute kidney injury, and low fibrinogen immediately narrowed the differential to a TMA process (Table 1 illustrates the trajectory of these abnormalities in relation to her neurologic decline). Within the spectrum of TMA, distinguishing among subtypes is critical yet challenging, as initial laboratory values can potentially overlap. TTP, an often idiopathic TMA caused by severe ADAMTS13 deficiency, classically presents with the pentad of MAHA, thrombocytopenia, fluctuating neurologic deficits, renal impairment, and fever [1]. Our patient had multiple strokes on imaging, encephalopathy and focal neurologic signs with laboratory values showing MAHA with thrombocytopenia acute kidney injury, raising TTP as a major concern. However, TTP usually lacks coagulopathy, as coagulation studies (PT/INR, fibrinogen) are typically normal in TTP [3]. In contrast, our patient’s prolonged INR and hypofibrinogenemia suggested DIC or DIC-like consumption. In fact, DIC and TTP can present simultaneously or be mistaken for each other in the clinical settings in the background of fast clinical deterioration [3]. DIC is an acquired syndrome of uncontrolled intravascular coagulation, often triggered by infection, malignancy, or shock, leading to both widespread microthrombi and bleeding [3]. In our patient with hypofibrinogenemia and coagulopathy (prolonged PT/INR) potentially indicating DIC-like consumption, then this case represents an uncommon and clinically consequential stroke phenotype of DIC. Another differential consideration was APS, an autoimmune hypercoagulable state known for both large-vessel and microvascular thromboses. APS frequently manifests with ischemic stroke, which is the most common arterial event, comprising 25% of APS-related events, and can feature livedo reticularis, thrombocytopenia, or hemolytic anemia [4]. In CAPS, a rapid thrombotic storm occurs with simultaneous clotting in multiple organs (often affecting small and large vessels over days) [4]. CAPS can mimic TTP/DIC with MAHA and consumptive coagulopathy, and it carries a very high mortality rate [4]. Our patient’s history of recurrent DVTs and strokes on therapeutic anticoagulation was indeed suspicious for APS; however, APS typically causes only mild thrombocytopenia (> 100 × 103/µL) and a positive Coombs test rather than frank MAHA [4]. No evidence of antiphospholipid antibodies was found in her immunologic workup, making APS less likely. We also evaluated atypical HUS, a complement-mediated TMA usually marked by predominant renal failure and hypertension. Atypical HUS can cause strokes and other organ damage, but typically there is an identifiable complement pathway mutation or precipitant (none evident here) and often severe hypertension (our patient’s blood pressure was not in malignant ranges) [3]. Secondary TMAs due to malignancy or malignant hypertension were also in the differential. Given the absence of a known inciting drug (negative urine toxicology) or malignant process, those were less favored. Finally, the patient’s diffuse encephalopathy and multifocal MRI lesions initially prompted consideration of central nervous system vasculitis or autoimmune encephalitis (e.g., N-methyl-D-aspartate (NMDA)-receptor encephalitis) (Fig. 1). Normal inflammatory markers and negative rheumatologic tests argued against primary vasculitis. The presence of clear ischemic infarcts (not just inflammatory lesions) without general systemic vasculitic processes and laboratory evidence of MAHA steered us away from primary inflammatory vasculitis. In summation, the leading diagnosis in this case was an acute TMA with features overlapping TTP and DIC, a situation reported in the literature where severe TTP can present with DIC-like labs [3]. One review estimates that while many TMA patients are initially mislabeled as having DIC, only 15% of DIC cases are true TMAs [3]. In our patient, interpretation of the ADAMTS13 activity (55%) warrants caution because the sample was drawn after transfusion of plasma products at the outside facility; passive replacement can transiently raise circulating ADAMTS13 and blunt a severe deficiency. Current British Society for Hematology guidelines emphasize banking a specimen before any plasma infusion and proceeding empirically when immune TTP is strongly suspected, rather than waiting for results [5]. Even when sampling is delayed, diagnostic yield persists: in a prospective cohort, ADAMTS13 activity remained < 10% in most immune TTP cases despite early PLEXs, with sensitivities of 89%, 83%, and 78% when testing was performed after the first, second, and third exchanges, respectively [6]. These data points show that a “normalized” post-transfusion result should not exclude TTP in the right clinical context, supporting empiric initiation of PLEX and corticosteroids when suspicion is high [5]. Furthermore, tools like the PLASMIC score can estimate the pretest probability of severe ADAMTS13 deficiency (immune TTP) and help triage urgent therapy while ADAMTS13 testing is pending [7]. In multi-center derivation/validation and subsequent external validation, scores 0 - 4, 5, and 6 - 7 correspond to low, intermediate, and high risk, respectively, with a sensitivity of 99% and negative predictive value (NPV) of 99% for ruling out immune TTP at a threshold ≥ 5 in suspected TMA cohorts [7]. Initial labs values after thrombectomy (platelets were 44 × 103/µL (0 points), hemolysis was not present via indirect bilirubin < 2 mg/dL (0), no active cancer (1), no transplant (1), MCV 80 - 82 fL (1), INR 2.6 - 3.6 (0), creatinine 2.2 - 3.5 mg/dL (0)) generated a PLASMIC score of 3/7 (low-risk category). Nonetheless, international guidelines emphasize that clinical judgment supersedes any score: obtain an ADAMTS13 sample before plasma products or PLEX when feasible, and if immune TTP remains strongly suspected, start PLEX ± caplacizumab/steroids without waiting for results [8]. This is significant because our patient’s ADAMTS13 assay (55%) was drawn after plasma infusion, and exogenous plasma can artifactually raise measured ADAMTS13 activity, potentially obscuring severe deficiency [9]. Thus, a low PLASMIC in the setting of overt MAHA or thrombocytopenia and neurologic injury should temper but not preclude empiric TTP-directed therapy when the bedside picture warrants it. The diagnostic ambiguity highlights a key point: when clinical suspicion for TMA is high, one should not await definitive tests to act, as delays can be deadly just like the catastrophic nature of our patient [1]. In practice, evidence of unexplained MAHA and thrombocytopenia, even without the full TTP pentad and normalized ADAMTS13, is sufficient to start treatment for TTP [1]. Unfortunately, in our case, the overlap of TMA subtypes confounded the clinical picture and contributed to a delay in targeted therapy.
Once a TMA is recognized as the culprit of stroke, management must pivot from standard stroke protocols to disease-specific therapies. Ischemic stroke in the context of TMA is not an ideal candidate for thrombolysis or routine antithrombotic strategies, due to both efficacy and safety concerns. In acute stroke care, intravenous thrombolysis (tissue-type plasminogen activator (tPA) or tenecteplase) is contraindicated in patients with active coagulopathy or platelets < 100 × 103/µL [10]. Our patient’s profound thrombocytopenia and hemorrhagic risk precluded thrombolytic use. There are reports of patients with unrecognized TTP who received tPA for stroke, often with poor results or delayed recovery due to the underlying TMA process continuing unchecked [11]. Antiplatelet therapy alone is also insufficient in TMA-related strokes. The microthrombi in TMA are formed by widespread platelet-vWF aggregation (in TTP) or fibrin deposition (in DIC) rather than the platelet activation pathways that aspirin or P2Y12 inhibitors target [12]. Thus, while antiplatelets are standard for secondary prevention in typical non-cardioembolic strokes, they do not address the root cause in TMA and may exacerbate bleeding tendencies [13]. For example, giving clopidogrel in an evolving TTP not only fails to stop thrombosis but has been associated with precipitating TTP in rare cases [13]. Similarly, anticoagulation is a double-edged sword in this scenario. In APS, long-term anticoagulation (warfarin, INR 2 - 3) is the cornerstone to prevent recurrent stroke, because clot formation is driven by antiphospholipid antibodies triggering the coagulation cascade [14]. However, indiscriminate anticoagulation in an undifferentiated TMA can be hazardous. For instance, if our patient actually has TTP or DIC with a high bleeding risk and ongoing PLEX, adding full-dose heparin could cause serious hemorrhage. In our case, the decision was made to hold anticoagulation initially due to the extensive infarction (risk of hemorrhagic transformation) and uncertainty of APS diagnosis. This illustrates the principle that therapy must be tailored to the specific TMA etiology, and when in doubt, treatments that address the TMA process (e.g., PLEX) should take priority over conventional antithrombotics [11].
The mainstay of treatment for TTP (or TTP-like microangiopathy) is urgent PLEX with replacement by fresh frozen plasma [12]. PLEX removes circulating ultra-large von Willebrand multimers and autoantibodies while replenishing ADAMTS13, thereby halting microvascular platelet clumping [12]. Outcomes in TTP have improved dramatically with this therapy, survival has risen from < 10-20% to > 80% in the PLEX era [12]. Best practice guidelines call for PLEX to begin as soon as possible, ideally within 4 - 8 h of suspicion, without waiting for ADAMTS13 assay confirmation [12]. In addition, high-dose corticosteroids are given to suppress any autoimmune driver [12]. In immune-mediated TTP, rituximab is often used to curb autoantibody production [12]. In refractory TTP, newly developed targeted therapy caplacizumab (an anti-vWF nanobody) can be added to prevent platelet-vWF interactions [12]. Caplacizumab in combination with PLEX has shown faster platelet count recovery and reduced TTP relapses in trials [12]. Our patient eventually received PLEX and steroids, but the delay (due to diagnostic uncertainty and concern for her renal failure) likely resulted in further thrombotic damage. Studies have shown that lesions in acute TTP are reversible with treatment, and follow-up imaging after PLEX may show improvement, suggesting that not all diffusion-weighted imaging (DWI) abnormalities represent permanent infarction [15]. This echoes the central challenge in managing atypical cases: one must sometimes treat empirically for TTP in the right clinical context, even when the diagnosis is clouded [1]. Experts emphasize that if TTP is in the differential, PLEX should be started promptly, because TTP untreated is rapidly fatal [1].
If an alternative TMA etiology emerges, therapy should be adjusted accordingly. For example, if CAPS were confirmed (e.g., by positive lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies), the treatment would center on anticoagulation and immunomodulation rather than PLEX alone. CAPS is typically managed with a combination of heparin, high-dose steroids, and often PLEX or intravenous immunoglobulin, plus treating any precipitating factors [4]. Similarly, an atypical HUS (complement-mediated) diagnosis would call for eculizumab, a terminal complement inhibitor, which has revolutionized atypical HUS outcomes in the past decade [16]. In patients with DIC secondary to sepsis or malignancy, the cornerstone is treating the underlying cause; supportive measures like blood product replacement are used, and in some cases low-dose heparin is cautiously employed if thrombosis dominates over bleeding [3]. Notably, platelet transfusions are avoided in TTP unless there is life-threatening hemorrhage, because more intravenous platelets can worsen microthrombosis [3]. Our patient’s management was a reflection of these nuances: we held platelet transfusions given concern for TTP, yet transfused cryoprecipitate and plasma to support coagulation.
Conclusions
In summary, this case highlights a clinically uncommon but deadly scenario of multifocal ischemic strokes caused by an underlying TMA. The overlap of TTP, DIC, and APS features created a diagnostic and therapeutic dilemma. The key lessons are to think beyond typical stroke etiologies when red flags like MAHA or thrombocytopenia appear, and to initiate TMA-directed treatment without delay when TTP or similar syndromes are suspected. Our patient suffered bilateral hemispheric strokes with cerebral edema by the time TMA therapy was instituted. Neurological injury in TTP can be severe and often fluctuates; stroke occurs in an estimated 8% of TTP presentations, and even after recovery, patients can have residual neurocognitive deficits [1, 12]. Standard stroke treatments (thrombolysis, antiplatelet agents) are often ineffective or contraindicated in these settings, and definitive therapy (PLEX, immunosuppression, or other targeted agents) should be prioritized [11]. It is therefore paramount to recognize TMA early, before irreversible organ damage accrues, as heightened awareness and rapid response to stroke-with-TMA cases can improve outcomes. When strokes occur as part of a TMA, aggressive identification and management of the underlying hematologic disorder is the only way to alter the otherwise catastrophic brain insult. This case reinforces the need for clinicians to remain vigilant for TMA in acute stroke and to act decisively, as saving brain tissue may ultimately depend on stopping the blood disorder that is attacking the microcirculation.
Acknowledgments
None to declare.
Financial Disclosure
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Written informed consent was obtained from the patient’s legal representative for publication of this case report and accompanying images.
Author Contributions
All authors contributed to the work reported in this manuscript. Taseal Ahmed, Yiorgos Antoniadis, Genesis Reyes Vega, Samir Ruxmohan were all involved in concept development, study design, definition of intellectual content, literature search, clinical and experimental studies, data acquisition, data analysis, statistical analysis, manuscript preparation, editing, review, and served as guarantor.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
APLS: antiphospholipid syndrome; DECT: dual-energy computed tomography; DIC: disseminated intravascular coagulation; ICA: internal carotid artery; MAHA: microangiopathic hemolytic anemia; NIHss: National Institutes of Health Stroke Scale; PLEX: plasma exchange; TMA: thrombotic microangiopathy; TTP: thrombotic thrombocytopenic purpura
| References | ▴Top |
- Jameie M, Heydari S, Ghabaee M, Amirifard H. Two ischemic stroke
events within 48 h: a case report of an unusual presentation of thrombotic thrombocytopenic
purpura. BMC Neurol. 2023;23(1):47.
doi pubmed - de Castro JTS, Appenzeller S, Colella MP, Yamaguti-Hayakawa G, Paula
EV, Annichinno-Bizzachi J, Cendes F, et al. Neurological manifestations in thrombotic
microangiopathy: Imaging features, risk factors and clinical course. PLoS One.
2022;17(9):e0272290.
doi pubmed - Wada H, Matsumoto T, Suzuki K, Imai H, Katayama N, Iba T, Matsumoto
M. Differences and similarities between disseminated intravascular coagulation and thrombotic
microangiopathy. Thromb J. 2018;16:14.
doi pubmed - Bahar Kelesoglu Dincer A, Erkan D. The ABCs of antiphospholipid
syndrome. Arch Rheumatol. 2023;38(2):163-173.
doi pubmed - Scully M, Rayment R, Clark A, Westwood JP, Cranfield T, Gooding R,
Bagot CN, et al. A British Society for Haematology Guideline: Diagnosis and management of
thrombotic thrombocytopenic purpura and thrombotic microangiopathies.
Br J Haematol. 2023;203(4):546-563.
doi pubmed - Wu N, Liu J, Yang S, Kellett ET, Cataland SR, Li H, Wu HM. Diagnostic
and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in
patients with acquired thrombotic thrombocytopenic purpura. Transfusion.
2015;55(1):18-24.
doi pubmed - Bendapudi PK, Hurwitz S, Fry A, Marques MB, Waldo SW, Li A, Sun L, et
al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with
thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4(4):e157-e164.
doi pubmed - Zheng XL, Vesely SK, Cataland SR, Coppo P, Geldziler B, Iorio A,
Matsumoto M, et al. ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura.
J Thromb Haemost. 2020;18(10):2486-2495.
doi pubmed - Papakonstantinou A, Kalmoukos P, Mpalaska A, Koravou EE, Gavriilaki
E. ADAMTS13 in the new era of TTP. Int J Mol Sci. 2024;25(15):8137.
doi pubmed - Demaerschalk BM, Kleindorfer DO, Adeoye OM, Demchuk AM, Fugate JE,
Grotta JC, Khalessi AA, et al. Scientific rationale for the inclusion and exclusion criteria for
intravenous alteplase in acute ischemic stroke: a statement for healthcare professionals from
the American Heart Association/American Stroke Association. Stroke. 2016;47(2):581-641.
doi pubmed - Taylor AO, Soares Ferreira Junior A, Dymm BL, Feng W, El Husseini N,
Onwuemene OA. Ischemic stroke in immune-mediated thrombotic thrombocytopenia purpura: diagnostic
and management challenges. Stroke. 2025;56(5):e144-e147.
doi pubmed - Hanlon A, Metjian A. Caplacizumab in adult patients with acquired
thrombotic thrombocytopenic purpura. Ther Adv Hematol. 2020;11:2040620720902904.
doi pubmed - Kirchner JT. Recognition of TTP is key when prescribing clopidogrel. AFP. 2000;62(12):2680.
- Ford B, Peela S, Roberts C. Secondary prevention of ischemic stroke:
updated guidelines from AHA/ASA. Am Fam Physician. 2022;105(1):99-102.
pubmed - Zhang Z, He M. Thrombotic thrombocytopenic purpura presenting as
stroke mimics with normal diffusion-weighted MRI. BMC Neurol. 2023;23(1):435.
doi pubmed - Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber
AO, Menne J, et al. Terminal complement inhibitor eculizumab in adult patients with atypical
hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis.
2016;68(1):84-93.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Neurology Research is published by Elmer Press Inc.